Idiopathic Pulmonary Fibrosis (IPF) is a disease that leads to respiratory failure and subsequent death. To prevent chronic lung diseases from rising further up the list of leading causes of death worldwide, new innovative therapeutic approaches are needed. This work aims to present a literature review on advances in regenerative therapies in the treatment of IPF. The bibliographic strategy of this research used papers published in last ten years taken from the electronic databases PubMed and Capes Periodical Portal, which include the current understanding of this pathology, and the different treatment modalities. The majority of selected articles were concentrated in the last three years of this review, corroborating the expectation of recent advancements given the current massive scientific research on IPF. This review focused on current treatments, their limitations and investigated cutting-edge research in regenerative therapies. The research resulted in the presentation of ongoing studies and therapies segregated between pharmacological manipulation and the use of stem cells. Both categories of treatment focus on restoring endogenous lung repair or targeting pathways that inhibit dysregulated regeneration. Genetic and epigenetic factors were constantly highlighted as extremely necessary for the diagnosis. Conclusively, given the heterogeneity of this pathology, the authors propose, for the next general IPF treatment protocol, the inclusion of a combination of therapies, accentuating the pro and post-installed disease components. Encouraging the established of specialized health centers is also part of the findings of this research. They promote close cooperation between pulmonologists, radiologists, biomedical geneticists, pathologists, among others.
| Published in | American Journal of Clinical and Experimental Medicine (Volume 13, Issue 4) |
| DOI | 10.11648/j.ajcem.20251304.15 |
| Page(s) | 99-134 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2025. Published by Science Publishing Group |
Idiopathic Pulmonary Fibrosis, Therapeutic Approach, Regenerative Therapies
TITLE | YEAR | COUNTRY | AUTHORS | OBJECTIVE | RESULTS | |
|---|---|---|---|---|---|---|
1 | Idiopathic Pulmonary Fibrosis: Review of Current Knowledge | 2024 | Slovakia | Muri, et al. | Presentation of ongoing stem cell trials | Mesenchymal stem cells have shown promising results; Research is underway for new options such as PRM-151, Pamrevlumab and Galectin-3 |
2 | Pulmonary fibrosis: Is stem cell therapy the way forward? | 2023 | Pakistan | Ikrama, et al. | Importance of stem cells in the treatment of IPF | Stem cells: regeneration and reduction of inflammation; Challenges: costs, ethical issues and immunological compatibility. |
3 | Stem cell-based therapy for pulmonary fibrosis | 2022 | China | Wenzhao Cheng, Yiming Zeng, Dachun Wang | Combination of stem cell therapies and gene therapy | Lung transplant: The only option for terminal stages; Stem cells: therapy for lung regeneration; |
4 | Therapeutic Applications of Mesenchymal Stem Cells in Idiopathic Pulmonary Fibrosis | 2021 | China | Yang, et al. | Presentation of ongoing stem cell trials | Mesenchymal stem cell (MSC) therapy regulates immunity and promotes tissue repair |
5 | Mesenchymal stem cells and pulmonary fibrosis: a bibliometric and visualization analysis of literature published between 2002 and 2021 | 2023 | China | Yang, et al. | Presentation of ongoing stem cell trials | Analysis of 1,457 publications on mesenchymal stem cells (MSCs) in the treatment of (IPF), increase in research in the area; USA led publications |
6 | Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review | 2019 | Japan | Tomonari Kinoshita, Taichiro Goto | Oncogenic panorama of IPF | Common molecular mechanisms between IPF and lung cancer; |
7 | The oncogenic landscape of the idiopathic pulmonary fibrosis: a narrative review | 2022 | Italy | Stella, et al. | Oncogenic panorama of IPF | Similarities of the molecular mechanisms of IPF with cancer; distinct dynamics; Personalized strategies. |
8 | Turning the tide: From fibrosis to regeneration following anti-fibrogenic cell vaccination | 2022 | United States of America | Ira Phadke, Alka Dwivedi, Naomi Taylor | Oncogenic panorama of IPF | Proposal for an immunotherapeutic vaccine targeting specific antigens of fibrogenic cells, such as ADAM12 in the liver and GLI1 in the lungs. Potential treatment for fibrotic diseases. |
9 | Lysophophatidic acid receptor 1 inhibition: a potential treatment target for pulmonary fibrosis | 2024 | United States of America | Volkmann et al. | Potential treatments | Investigation of ATX and LPAR1 Inhibitors as Promising Therapies in Clinical Trials for the Treatment of the Disease |
10 | Targeting Alveolar Repair in Idiopathic Pulmonary Fibrosis | 2021 | United Kingdom | Ptasinski VA, Stegmayr J, Belvisi MG, Wagner DE, Murray LA. | Potential treatments | Stem cell modulation; anti-inflammatory and antifibrotic therapies; the body's own repair capacity; promotion of tissue regeneration without stimulating the formation of pathological scars; understanding the molecular mechanisms that regulate alveolar repair; identification of therapeutic targets improving the prognosis of patients with IPF. |
11 | Target tumour suppressor p53 for organ fibrosis therapy | 2024 | China | Bao, et al. | Potential treatments | Modifying p53 activity is a promising therapeutic approach to treat IPF |
12 | Idiopathic pulmonary fibrosis and the role of genetics in the era of precision medicine | 2023 | Spain | Gonzalez, et al. | Genetic sequencing (protocol) | Explores how rare and common genetic variants influence the risk and progression of idiopathic pulmonary fibrosis (IPF). It also discusses how genomic technologies can improve diagnosis, prognosis, and genetic screening strategies for IPF. |
13 | The Inflammasome NLR Family Pyrin Domain-Containing Protein 3 (NLRP3) as a Novel Therapeutic Target for Idiopathic Pulmonary Fibrosis | 2022 | United States of America | Biancatell, et al. | Potential treatments | Reviews evidence on NLRP3 activation in idiopathic pulmonary fibrosis and highlights recent advances in direct and indirect NLRP3 inhibitors. |
14 | The Genetic and Epigenetic Footprint in Idiopathic Pulmonary Fibrosis and Familial Pulmonary Fibrosis: A state-of-the-Art Review | 2022 | Italy | Tirelli, et al. | Epigenetic factors of IPF such as biomarkers or druggable targets | Analyzes the genetic and epigenetic mechanisms involved in idiopathic pulmonary fibrosis (IPF) and familial pulmonary fibrosis (FPF). Highlights mutations in genes related to telomerase (TERT, TERC, PARN, RTEL1) and surfactant proteins (SFTPC, SFTPA1, SFTPA2, ABCA3) as well as polymorphism in the MUC5B and TOLLIP genes, which contribute to fibrogenesis. The study also addresses epigenetic alterations such as DNA methylation. |
15 | European Respiratory Society statement on familial pulmonary fibrosis | 2022 | France | Borie, et al. | Genetic sequencing (protocol) | Addresses genetic predisposition in IPF evidenced by several genetic mutations associated with the disease. The statement provides guidelines on genetic sequencing, clinical counseling, management and screening of patients with familial pulmonary fibrosis (FPF) and their family members, aiming to improve the diagnosis and treatment of this condition. |
16 | Genetic Risk Factors for Idiopathic Pulmonary Fibrosis: Insights into Immunopathogenesis | 2020 | United States of America | Jacob E Michalski, David A Schwartz | Genetic factors involved in IPF | It explores the genetic factors associated with the development of IPF and their implications in the immunopathogenesis of the disease. The authors highlight that genetic variations account for at least one third of the risk of developing IPF. Among these, the gain-of-function variant in the promoter of the MUC5B gene is indicated as the most significant risk factor, genetic or otherwise, for the disease. |
17 | Revealing the Pathogenic and Aging-related Mechanisms of the Enigmatic Idiopathic Pulmonary Fibrosis | 2014 | Mexico | Moisés Selman, Annie Pardo | Genetic variants in the aged lung | Explores the pathogenetic and age-related mechanisms of idiopathic pulmonary fibrosis (IPF). The authors propose that aberrant activation of alveolar epithelial cells and fibroblasts in aged lungs plays a crucial role in the pathogenesis of IPF |
18 | Precision medicine advances in idiopathic pulmonary fibrosis | 2023 | United States of America | Karampitsakos, et al. | Precision medicine in IPF patients | The work emphasizes that the adoption of precision medicine strategies in IPF has the potential to transform disease management but requires continued effort to integrate these scientific discoveries into daily clinical practice. |
19 | Molecular and Genetic Biomarkers in Idiopathic Pulmonary Fibrosis: Where Are We Now? | 2023 | Greck | Tomos, et al. | Epigenetic factors of IPF such as biomarkers or druggable targets | The article highlights the relevance of molecular and genetic biomarkers in understanding and managing IPF, emphasizing the need for further research to validate and implement these biomarkers in clinical practice. |
20 | Research progress in the molecular mechanisms, therapeutic targets, ad drugs development of Idiopathic Pulmonary Fibrosis | 2022 | China | Ma, et al | Genetic factors involved in IPF | This article summarizes advances in understanding the molecular mechanisms of IPF and how these discoveries are being translated into new therapeutic strategies and drug development aimed at improving management and clinical outcomes for patients. |
21 | Pharmacological expansion of type 2 alveolar epithelial cells promotes regenerative lower air repair | 2024 | United States of America | Shao, et al. | Potential treatments | This article suggests that DPP4 inhibition may be a promising strategy to stimulate lung regeneration in diseases such as idiopathic pulmonary fibrosis (IPF) offers a new therapeutic approach for degenerative lung conditions. |
22 | US patent covers brilaroxazine in treating IPF, like diseases | 2024 | United States of America | Bhat; Laxminarayan | Potential treatments | In 2024, the United States Patent and Trademark Office (USPTO) granted patent No. 12053477 to Reviva Pharmaceuticals Holdings for the use of brilaroxazine in the treatment of idiopathic pulmonary fibrosis (IPF) and other fibrosing lung diseases, including those associated with chronic obstructive pulmonary disease (COPD), sickle cell anemia, scleroderma, and lung cancer. |
23 | Idiopathic Pulmonary Fibrosis: Addressing the current and future therapeutic advances along with the role of Sotatercept in the management of pulmonary hypertension | 2023 | India | Hadi, et al. | Potential treatments | The article provides valuable insights into current and future therapeutic options for IPF, emphasizing the need for continued research to improve patient outcomes. |
24 | Promising advances in treatments for the management of idiopathic pulmonary fibrosis | 2024 | Italy | Carmelo Sofia, Alessia Comes, Giacomo Sgalla, Luca Richeldi | Potential treatments | This multifaceted approach is expected to significantly improve the overall treatment of patients with IPF, supporting optimistic expectations of reversing fibrotic changes and ultimately restoring lung function to a normal state. |
25 | A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models | 2024 | China | Feng Ren, et al. | Epigenetic factors of IPF such as biomarkers or druggable targets | A generative AI platform was used to unbiasedly identify TNIK as an antifibrotic target. TNIK has also been implicated in multiple hallmarks of aging, suggesting broader therapeutic potential |
26 | Exploring Therapeutic targets for molecular therapy of idiopathic pulmonary fibrosis | 2024 | China | Li, et al. | Genetic factors involved in IPF | It has mainly focused on therapeutic targets that have not only entered clinical trials but have also been publicly published with their clinical results. Until now, many researchers have focused on the downstream effectors of gene expression, such as cytokines and enzymes. Epigenetic modification also plays a vital role in IPF. |
27 | Current and Novel Treatment Modalities of Idiopathic Pulmonary Fibrosis | 2024 | United States of America | Mahnoor Arshad, Zoraize Moeez Athar, Tasneem Hiba | Current treatment modalities | This article provides an overview of current treatment modalities for IPF and explores the need for new therapeutic approaches. |
28 | The Bright side of fibroblastic: molecular signature and regenerative cues in major organs | 2021 | Portugal | Rita N Gomes, Filipa Manuel, Diana S Nascimento | Potential treatments | The positive side of fibroblasts: molecular signature and regenerative signals in major organs |
29 | Human Adipose-derived Mesenchymal Stem Cells Attenuate Early Stage of Bleomycin Induced Pulmonary Fibrosis: Comparison with Pirfenidone | 2016 | India | Reddy, et al. | Presentation of ongoing stem cell trials | In the present study, AD-MSCs were shown to be effective in improving the symptoms of pulmonary fibrosis, increasing survival capacity and protecting against pulmonary fibrosis comparatively better than pirfenidone. |
30 | Engineered Lung Tissues Prepared from Decellularized lung slices | 2022 | United States of America | Leiby, et al. | Precision medicine in IPF patients | In summary, this protocol describes a robust system to generate engineered lung tissues for culture studies of AEC2s, fibroblasts, and endothelial cells within acellular ECM lung slice scaffolds. |
31 | Early decrease in erector spinae muscle area and future risk of mortality in idiopathic pulmonary fibrosis | 2020 | Japan | Nakano, et al. | Epigenetic factors of IPF such as biomarkers or druggable targets | Early decrease in ESMCSA may be a useful predictor of prognosis in patients with IPF. |
32 | Innovative Pre-Clinical Data Using Peptides to Intervene in the Evolution of Pulmonary Fibrosis | 2023 | Brazil | Simon, et al. | Potential treatments | This work aimed to apply a therapeutic alternative using immunomodulatory peptides in a murine model of chronic pulmonary fibrosis. |
33 | Administration of Collagen Peptide Prevents the Progression of Pulmonary Fibrosis in Bleomycin-Treated Mice | 2023 | Japan | Yoshihara, et al. | Potential treatments | The results of this study suggest that CP (PEPTIDE COLLAGEN) supplementation prevents the development of IPF by acting as an anti-inflammatory agent. |
Mediators | IPF | Lung Cancer | |
|---|---|---|---|
Abnormal mRNA | let-7 | down-regulated | down-regulated |
miR-21 | up-regulated | up-regulated | |
miR-29 | down-regulated | down-regulated | |
miR-30 | down-regulated | down-regulated | |
miR-155 | up-regulated | up-regulated | |
miR-200 | down-regulated | down-regulated | |
Cell-free DNA | - | up-regulated | up-regulated |
Glycoprotein | Thy-1 | down-regulated | down-regulated |
Connexin | Cx43 | down-regulated | down-regulated |
Growth Factors | TGF-β | up-regulated | up-regulated |
PDGF | up-regulated | up-regulated | |
Migration | VEGF | up-regulated | up-regulated |
FGF | up-regulated | up-regulated | |
laminin | up-regulated | up-regulated | |
fascin | up-regulated | up-regulated | |
Pathways | heat shock protein 27 | up-regulated | up-regulated |
Wnt pathway | up-regulated | up-regulated | |
PI3K/Akt pathway | up-regulated | up-regulated | |
Immune Cells | FAM | up-regulated | up-regulated |
MDSC | up-regulated | up-regulated | |
Treg | down-regulated | up-regulated | |
Gene 1 | Main Function | Profibrotic Mechanism |
|---|---|---|
TERT | Telomerase | Decreased activity of telomerase |
TERC | Reverse Transcription in Telomerase | Decreased activity of telomerase |
PARN | Stability of mRNA in Telomerase | Shortening of telomeres |
RTEL1 | DNA helicase in Telomerase | Shortening of telomeres |
SFTPA1 | Modulate surface tension in the alveoli | Increased ER stress |
SFTPA2 | Modulate innate and adaptive immunity | Increased ER stress |
SFTPC | Stabilize the surfactant fluid | Increased ER stress |
ABCA3 | Lipid transport across membranes | Increased ER stress and apoptosis |
MUC5B | Mucin 5B production | Muco-ciliary disfunction |
TOLLIP | Inhibitory adaptor protein within TLR | Decreased protection against ROS |
Treatment Modality | Description |
|---|---|
Pirfenidone | Oral medication that reduces fibroblast activity and pro-inflammatory cytokines. |
Nintedanibe | Oral medication that inhibits multiple tyrosine kinases involved in fibroblast proliferation. |
Oxygen Therapy | It provides supplemental oxygen to improve oxygenation and relieve symptoms of hypoxemia. |
Pulmonary Rehabilitation | Comprehensive programs, including exercise training, education, and support to improve lung function and overall well-being. |
Lung Transplantation | Considered for patients with advanced idiopathic pulmonary fibrosis who meet specific criteria to improve survival and quality of life. |
Treatment | Mechanism of action | Clinical effects |
|---|---|---|
Current therapies | ||
Pirfenidone | Antifibrotic and anti-inflammatory | Slows rate of decline in FVC |
Nintedanibe | Antifibrotic and anti-inflammatory | Slows rate of decline in FVC |
Oral corticosteroids, opioids | Antitussive | Decreases cough and improves quality of life |
Anti-acids, proton pump inhibitors | Decreases GERD | Benefits unclear |
Lung transplantation | Surgical replacement of one or both lungs | Available only as a potentially curative therapy |
Therapies in development | ||
PRM-151 | Recombinant human pentraxin-2; acts as an antifibrotic agent | Slows rate of decline in FVC |
Pamrevlumab | Fully human recombinant monoclonal antibody to CTGF | Slows rate of decline in FVC |
TD139 | Small molecule inhibitor of galectin-3 | Decreases plasma biomarkers of inflammation; study in progress to assess effect on FVC |
PLN-74809 | Blocks activation of the TGFβ pathway | Study in progress with end-points of safety, tolerability, and pharmacokinetics |
TRK-250 | Suppresses TGFβ expression | Study in progress to assess the safety and tolerability of single and multiple inhaled doses |
TITLE | TARGET OR FACTOR ASSOCIATED WITH IPF IDENTIFIED | THERAPY OR MEDICATION |
|---|---|---|
Lysophophatidic acid receptor 1 inhibition: a potential treatment target for pulmonary fibrosis | Lysophosphatidic acid (LPA)-mediated activation of LPA receptor 1 (LPAR1); and the LPA-producing enzyme autotaxin (ATX) and activation of LPAR1 | Signaling in pulmonary fibrosis and to help differentiate new inhibitors in development |
Targeting Alveolar Repair in Idiopathic Pulmonary Fibrosis | reparative epithelial progenitor cells in the alveolar region of the lung | Endogenous alveolar repair |
The Bright side of fibroblastic: molecular signature and regenerative cues in major organs | The bright side of fibroblasts: molecular signature and regenerative signals in major organs: updated overview of fibroblast-derived regenerative signaling in different organs and discuss how this capacity may be compromised with aging | We also introduce a new paradigm for regenerative therapies based on the reversion of adult fibroblasts to a fetal/neonatal-like phenotype. |
Target tumor suppressor p53 for organ fibrosis therapy | Tumor suppressor p53 (p53), known for its regulatory role in cell proliferation, apoptosis, aging and metabolism in various tissues | Development of strategies targeting p53 for the treatment of organ fibrosis |
The Inflammasome NLR Family Pyrin Domain-Containing Protein 3 (NLRP3) as a Novel Therapeutic Target for Idiopathic Pulmonary Fibrosis | NLR family pyrin domain-containing protein 3 (NLRP3), once activated, promotes the production of IL-1β, IL-18, and innate immune responses. | Inhibition of NLRPP3 may offer a promising approach against the fibrotic process in the lungs |
Pharmacological expansion of type 2 alveolar epithelial cells promotes regenerative lower air repair | Dipeptidyl peptidase 4 (DPP4) inhibitors selectively expand AEC2s (alveolar epithelial cells type 2 are stem cells in the adult lung that contribute to lower airway repair) and are broadly effective in several mouse models of lung injury. | Z-97, a persistent, locally administered pulmonary DPP4 inhibitor that broadly promotes efficacy in mouse lung injury models with minimal peripheral exposure and good tolerability. |
US patent (US-12053477-B2) covers brilaroxazine in treating IPF, like diseases | Dopamine and serotonin play important roles in regulating processes such as fibrosis (scarring) and inflammation. | Brilaroxazine is a new chemical entity designed to modulate the activity of dopamine and serotonin receptors, two key signaling molecules |
Idiopathic Pulmonary Fibrosis: Addressing the current and future therapeutic advances along with the role of Sotatercept in the management of pulmonary hypertension | Inhibition of several tyrosine kinases decreases the proliferative activities that lead to fibrosis. | Saracatinib is a tyrosine kinase inhibitor that primarily targets the SRC protein, potentially used to treat diseases such as pulmonary fibrosis by inhibiting fibroblast activity and collagen deposition, while Sotatercept is a fusion protein used to treat pulmonary arterial hypertension (PAH) by targeting specific pathways that contribute to abnormal blood vessel growth in the lungs; |
Promising advances in treatments for the management of idiopathic pulmonary fibrosis | Presents a collection of phase 3 tests: Targets: PDe4B; Fibroblast activities; CTGF; Innate immune response; Lung microbiome | "BI 1,015,550; Inhaled treprostinil; Pamrevlumab |
Innovative Pre-Clinical Data Using Peptides to Intervene in the Evolution of Pulmonary Fibrosis | Immunomodulatory peptides ToAP3 and ToAP4 | Immunomodulatory peptides ToAP3 and ToAP4: both peptides controlled experimental IPF, maintaining tissue characteristics and standard functional properties and regulating the production of cytokines associated with fibrosis. The data obtained in this work show that the regulation of the immune response by ToAP3 and ToAP4 can control the alterations that cause the fibrotic process, making both peptides potential therapeutic alternatives and/or adjuvants for IPF. |
Administration of Collagen Peptide Prevents the Progression of Pulmonary Fibrosis in Bleomycin-Treated Mice | Peptide collagen | The present study showed that administration of peptide collagen derived from chicken feet suppressed the progression of Pulmonary Fibrosis in mice. |
Exploring Therapeutic targets for molecular therapy of idiopathic pulmonary fibrosis | Transforming growth factor beta, vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor, lysophosphatidic acid, interleukin-13, Rho-associated coiled-coil forming protein kinase family, and Janus kinases/signal transducers and activators of transcription pathway. | Transforming growth factor beta, vascular endothelial growth factor, platelet-derived growth factor, fibroblast growth factor, lysophosphatidic acid, interleukin-13, Rho-associated coiled-coil forming protein kinase family, and Janus kinases/signal transducers and activators of transcription pathway. |
Turning the tide: From fibrosis to regeneration following anti-fibrogenic cell vaccination | ADAM12, a member of the A Disintegrin and Metalloprotease (ADAM) family of proteins that harbors extracellular metalloprotease and intracellular signaling properties. ADAM12 is an interesting target because tissue-resident ADAM12+ cells have been identified as the source of most a-SMA+ myofibroblasts generated after acute muscle and dermal injury, and its expression is largely limited to the embryonic period. GLI1 is expressed in mesenchymal cells after injury, giving rise to a-SMA+ myofibroblasts after tissue injury | Novel immunotherapeutic vaccine approach to target fibrotic lesions in models of liver and lung fibrosis: (This therapeutic strategy is similar to a cancer vaccine approach, but instead of targeting tumor-specific antigens, it targets fibrogenic cell-specific antigens.) Vaccination with a lentiviral vector encoding Gli1 abrogates bleomycin-induced lung fibrosis |
A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models | It was identified TRAF2- and NCK-interacting kinase (TNIK) as an antifibrotic target using a predictive artificial intelligence (AI) approach. | "INS018_055, a small molecule TNIK inhibitor, was generated, which exhibits desirable drug-like properties and antifibrotic activity in different organs in vivo via oral, inhaled or topical administration. INS018_055 possesses anti-inflammatory effects in addition to its antifibrotic profile, validated in multiple in vivo studies. |
Target molecule | Drug or treatment | Administration route | Clinical trial stage | Research type | Patients number | Outcomes |
|---|---|---|---|---|---|---|
galectin-3 | TD139, a galectin-3 inhibitor | Inhalation, 0.3, 3, or 10 mg for 14 days | I/IIa | Double blinded, Randomized | 24 patients. Each cohort: TD139 (n = 5) or placebo (n = 3) | Inhibition of Gal-3 expression in the lung was associated with reductions in plasma biomarkers (TD139 groups compared with placebo). |
αvβ6 integrin | BG00011, a humanized monoclonal antibody against αvβ6 integrin | Subcutaneous, 56 mg once weekly | IIb | Double blinded, Randomized | 106 patients. BG00011 (n = 54) or placebo (n = 52) | No significant difference in FVC change from baseline between patients who received BG00011 or placebo at week 26. |
CTGF | Pamrevlumab, a recombinant human antibody that binds to CTGF | Intravenous, 30 mg/kg, every 3 weeks over 48 weeks | III | Double blinded, Randomized | 356 patients. | Study did not meet its primary endpoint. |
LPA receptor | BMS-986020 (Bristol-Myers Squibb), an antagonist of the lysophosphatidic acid (LPA) receptor 1 (LPA1) | Oral, 600 mg, once/twice daily for 26 weeks | II | Double blinded, Randomized | 143 patients. placebo (n = 47), 600 mg qd (n = 48), 600 mg bid (n = 48) | Patients treated with BMS-986020 experienced a significantly slower rate of decline vs placebo in FVC at week 26. |
Autotaxin (the primary enzyme responsible for the production of LPA) | GLPG1690 (Galapagos, Mechelen, Belgium), a selective inhibitor of autotaxin | Oral, 600 mg/200 mg once daily for at least 52 weeks | III | Double blinded, Randomized | 1306 patients. | GLPG1690 did not improve clinical outcomes compared with placebo. |
IL-13 | Lebrikizumab, a monoclonal antibody inhibits the secretion of IL-13 | Subcutaneous, 250 mg, every 4 weeks | II | Double blinded, Randomized | 505 patients. In cohort A, 154 patients, lebrikizumab (n = 78) or placebo (n = 76). In cohort B, 351 patients received pirfenidone, lebrikizumab (n = 174) or placebo (n = 177) | The predicted decline of the annualized rate of FVC% was not met. |
IL-4 and IL-13 | SAR156597, a monoclonal antibody targeting IL-4 and IL-13 | Subcutaneous, 200 mg once a week or once every 2 weeks | IIb | Double blinded, Randomized | 325 patients, placebo (n = 109), SAR156597 Q2W (n = 108), SAR156597 QW (n = 108) | SAR156597 failed to demonstrate benefit in treating IPF patients. |
Rho-associated protein kinase 2 (ROCK2) | KD025 (or SLX-2119; Kadmon Corporation, Belumosudil), a selective ROCK2 inhibitor | Oral, 400 mg once a week | II | Open-label, Randomized | 76 patients. KD025 (n = 52) or best supportive care (n = 24) | KD025 reduced the decline of FVC by 73% versus patients receiving supportive care at week 24. |
Pulmonary disease marker | Findings in screening asymptomatic relatives | Remarks from Task Force |
|---|---|---|
Physical examination, symptoms | ||
Dyspnea/MRC-5 | May be present; not predictive for pre-clinical pulmonary fibrosis | |
Frequent cough | May be present; not predictive for pre-clinical pulmonary fibrosis | |
Clubbing | May be present | |
Inspiratory crackles | May be present | |
Pulmonary function | Can be performed in subjects from age 6 years onward | |
FVC | Below normal values rare | |
DLCO | Below normal values may be present; lower in subjects with ILD changes than without ILD changes on HRCT | |
TLC | Below normal values rare; lower in subjects with ILD changes than without ILD changes on HRCT | |
Radiology HRCT | 14–25% have ILD changes on HRCT or a diagnosis of ILD on first screening visit | One-third of SFTPA1/SFTPA2 mutation carriers may develop lung cancer |
Genetic sequencing | Disease-causing variants in TRGs and SRGs | In families with a (likely) pathogenic SRG mutation: family members without the mutation are not at increased risk for FPF; in families with a (likely) pathogenic TRG mutation: family members without the mutation may have inherited short telomeres and may be at risk of development of STS |
Extrapulmonary signs and symptoms of STS | In all families with unknown cause or TRG mutation | |
Hematological markers | Mean corpuscular volume above normal; red blood cells or platelets below normal | Abnormalities in one or several hematological cell lines are a sign of marrow dysfunction in asymptomatic carriers of TRG mutations |
Liver enzymes | Elevated liver enzymes | Abnormalities in liver enzymes are a sign of hepatic disease in asymptomatic carriers of TRG mutations |
Hair greying | Before 30 years of age | No consensual definition |
Telomere length | <10th percentile | Low values for age associate with marrow dysfunction in asymptomatic carriers of TRG mutations |
Biomarker | Pathogenetic Process | Diagnostic | Prognostic | Specimen- |
|---|---|---|---|---|
S100A4 | Fibrogenesis | ++ | ++ | Serum BALF Lung tissue |
cCK-18 | AECs apoptosis | + | - | Serum |
KL-6 | Alveolar epithelial marker | + | +++ | Blood BALF |
YKL-40 | Adhesion molecule | - | ++ | Blood BALF Lung tissue |
MMP-7 | Extracellular remodelling | ++ | +++ | Blood BALF Lung tissue |
ICAM1 | Adhesion molecule | - | ++ | Serum Blood |
SPA & SPD | Alveolar epithelial markers | - | ++ | Serum |
LOXL2 | Extracellular matrix remodelling Fibrogenesis | - | + | Serum Lung tissue |
Periostin | Extracellular matrix remodelling Fibrogenesis | - | + | Serum Lung tissue |
CCL-18 | Alternative alveolar macrophage activation | - | + | Serum BALF |
IL-8 | Potent chemotactic activity for polymorphonuclear leukocytes | - | + | Blood BALF |
OPN | Inflammation Fibroblast migration and proliferation | - | + | Serum BALF Lung tissue |
LC3β | Autophagy | - | + | Lung Tissue |
BiP, XBP1 | Unfolded protein response | - | + | Lung Tissue |
IPF | Idiopathic Pulmonary Fibrosis |
REC | Research Ethics Committee |
FPF | Familial Pulmonary Fibrosis |
ESCs | Embryonic Stem Cells |
iPSCs | Induced Pluripotent Stem Cells |
MSCs | Mesenchymal Stem Cells |
EHC | Specialized Health Centers |
ILD | Interstitial Lung Disease |
| [1] |
AFYA BLOG GRADUATION. Regenerative Medicine: what it is and how it works? Brazil, Available at:
https://graduacao.afya.com.br/medicina/medicina-regenerativa-o-que-e Accessed on: 11 June. 2024. |
| [2] | ALONSO-GONZALEZ, A. et al. Idiopathic pulmonary fibrosis and the role of genetics in the era of precision medicine. Front Med (Lausanne), Spain, v. 27, n. 10, p. 1152211, abr./2023. Available at: |
| [3] |
AMARAL, Alexandre Franco; COLARES, P. D. F. B; KAIRALLA, Ronaldo Adib. Idiopathic Pulmonary Fibrosis: Current Diagnosis and Treatment. Brazilian Pneumological Journal, Brazil, v. 49, n. 4, p. 20230085, Aug./2023. Available at:
https://jornaldepneumologia.com.br/details/3847/en-US/idiopathic-pulmonary-fibrosis--current-diagnosis-and-treatment (Accessed on: 8 July 2024). |
| [4] | ARSHAD, Mahnoor; ATHAR, Zoraize Moeez; HIBA, Tasneem. Current and Novel Treatment Modalities of Idiopathic Pulmonary Fibrosis. Cureus, USA, v. 16, n. 3, p. 56140, March. /2024. Available at: |
| [5] | BADDINI-MARTINEZ, J. et al. Update on the Diagnosis and Treatment of Idiopathic Pulmonary Fibrosis. Brazilian Pneumological Journal, Brazil, v. 41, n. 5, p. 454-466, Aug. 2015. |
| [6] | BAO, Y. et al. Targeting tumor suppressor p53 for organ fibrosis therapy. Cell Death Dis, China, v. 15, n. 5, p. 336, May/2024. Available at: |
| [7] | BHAT, Laxminarayan. Methods for treating pulmonary fibrosis.: Revive Pharmaceuticals, Inc. (Cupertino, CA). n. USA 20230061592 A1. Deposit 28 March. 2022. Concession: 02 March. 2023) Evaluation of Brilaroxazine (RP5063) in a Bleomycin-Induced Rodent Model of Idio-pathic Pulmonary Fibrosis. |
| [8] | BIANCATELL, R. M. L. C; SOLOPOV, Pavel A; CATRAVAS, John D. The Inflammasome NLR Family Pyrin Domain-Containing Protein 3 (NLRP3) as a Novel Therapeutic Target for Idiopathic Pulmonary Fibrosis. Am J Patho, USA, v. 192, n. 6, p. 837-846, jun./2022. Available at: |
| [9] | BORIE, R. et al. European Respiratory Society statement on familial pulmonary fibrosis. European Respir J, France, v. 61, n. 3, p. 2201383, March /2023. Available at: 2025) |
| [10] | BUENO, M. et al. Mitochondria Dysfunction and Metabolic Reprogramming as Drivers of Idiopathic Pulmonary Fibrosis. Elsevier-Redox Biology, USA, v. 33, n. 101509, p. 32234292, March /2020. Available at: |
| [11] | CHENG, Wenzhao; ZENG, Yiming; WANG, Dachun. Stem cell-based therapy for pulmonary fibrosis. Stem Cell Therapy, China, v. 13, n. 1, p. 492, Oct./2022. Available at: |
| [12] | GOMES, Rita N; MANUEL, Filipa; NASCIMENTO, Diana S. The bright side of fibroblasts: molecular signature and regenerative cues in major organs. NPJ Regen Med, Portugal, v. 6, n. 1, p. 43, Aug./2021. Available at: |
| [13] | HADI, D. D. et al. Idiopathic pulmonary fibrosis: Addressing the current and future therapeutic advances along with the role of Sotatercept in the management of pulmonary hypertension. Immun Inflamm Dis, India, v. 11, n. 11, p. 1079, Nov./2023. Available at: |
| [14] | IKRAMA, M. et al. Pulmonary fibrosis: Is stem cell therapy the way forward? J Taibah Univ Med Sci, Pakistan, v. 19, n. 1, p. 82-89, Oct./2023. Available at: |
| [15] | KARAMPITSAKOS, T. et al. Precision medicine advances in idiopathic pulmonary fibrosis. Ebio Medicine, USA, v. 95, n. 1, p. 104766, Sept./2023. Available at: |
| [16] |
KINOSHITA, Tomonari; GOTO, Taichiro. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int J Mol Sci., Japan, v. 20, n. 6, p. 1461, May/2019. Available at:
https://doi.org/10.3390/ijms20061461 (Accessed on: 18 June. 2024). |
| [17] | LECHNER, A. J.; MATUSCHAK, G. M.; BRINK, D. S. Lungs: An Integrated Approach to Disease. 1. ed. Brazil, Lange, 2013. p. 1-452. |
| [18] | LEIBY, K. L. et al. Engineered Lung Tissues Prepared from Decellularized Lung Slices. J Vis Exp, USA, v. 21, n. 179, p. 179, Jan./2022. Available at: |
| [19] | LEMOS, Carolina; RAOTA, T. M.; V. New Pharmacological Perspectives for the Treatment of Idiopathic Pulmonary Fibrosis: An Integrative Literature Review. Brazilian Medical Students Journal, Brazil, v. 8, n. 11, p. 352, Aug. 2023. |
| [20] | LI, Y. et al. Exploring therapeutic targets for molecular therapy of idiopathic pulmonary fibrosis. Sci Prog, China, v. 107, n. 2, p. 368504241247402, April/2024. Available at: |
| [21] | MA, H. et al. Research Progress in the Molecular Mechanisms, Therapeutic Targets, and Drug Development of Idiopathic Pulmonary Fibrosis. Front Pharmocol, China, v. 21, n. 13, p. 963054, July/2022. Available at: |
| [22] | MEI, Q. et al. Idiopathic Pulmonary Fibrosis: An Update on Pathogenesis. Frontiers in Pharmacology, China, v. 19, n. 12, p. 797292, Jan./2022. Available at: |
| [23] | MICHALSKI, Jacob E; SCHWARTZ, David A. Genetic Risk Factors for Idiopathic Pulmonary Fibrosis: Insights into Immunopathogenesis. J Inflammatory Res, USA, v. 5, n. 13, p. 1305, Jan./2021. Available at: |
| [24] | MURI, J. et al. Idiopathic Pulmonary Fibrosis: Review of Current Knowledge. Physiological Research, Slovakia, v. 73, n. 4, p. 487-497, March/2024. Available at: |
| [25] | NAKANO, A. et al. Early decrease in erector spinae muscle area and future risk of mortality in idiopathic pulmonary fibrosis. Sci Rep, Japan, v. 10, n. 1, p. 2312, Feb./2020. Available at: |
| [26] | OLDHAM, Justin M; VANCHERI, Carlo. Rethinking Idiopathic Pulmonary Fibrosis. Clinics in Chest Medicine, USA, v. 42, n. 2, p. 263-273, June/2021. Available at: |
| [27] | PHADKE, Ira; DWIVEDI, Alka; TAYLOR, Naomi. Turning the tide: From fibrosis to regeneration following anti-fibrogenic cell vaccination. Cell Stem Cell, USA, v. 29, n. 10, p. 1421-1423, Oct. 2022. |
| [28] | PTASINSKI, V. A. et al. Targeting Alveolar Repair in Idiopathic Pulmonary Fibrosis. Am J Respir Cell Mol Biol, England, v. 65, n. 4, p. 347-365, Oct./2021. Available at: |
| [29] | REDDY, M. et al. Human Adipose-derived Mesenchymal Stem Cells Attenuate Early Stage of Bleomycin Induced Pulmonary Fibrosis: Com-parison with Pirfenidone. International Journal of Stem Cells, India, v. 9, n. 2, p. 192-206, Nov./2016. Available at: |
| [30] | REN, F. et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat Biotechnology, China, v. 43, n. 1, p. 63-75, March/2024. Available at: |
| [31] | SELMAN, Moisés; PARDO, Annie. Revealing the pathogenic and aging-related mechanisms of the enigmatic idiopathic pulmonary fibrosis. an integral model. Am J Respir Crit Care Med., Mexico, v. 189, n. 10, p. 1161-1172, May/2014. Available at: |
| [32] | SGALLA, G. et al. Idiopathic Pulmonary Fibrosis: Pathogenesis and Management. Respiratory Research, Italy, v. 19, n. 1, p. 32, Feb. 2019. Available at: |
| [33] | SHAO, S. et al. Pharmacological expansion of type 2 alveolar epithelial cells promotes regenerative lower airway repair. Proc Natl Academy Sci USA, v. 121, n. 6, p. 2400077121, April/2024. Available at: |
| [34] | SILVA et al. Challenges and Perspectives in the Management of Idiopathic Pulmonary Fibrosis: Towards More Effective Therapies. Ibero-America Magazine of Humanities and Science Education, Brazil, v. 10, n. 4, p. 1153-1160, April/2024. Available at:) |
| [35] | SIMON, K. S. et al. Innovative Pre-Clinical Data Using Peptides to Intervene in the Evolution of Pulmonary Fibrosis. Int J Mol Sci, Brazil, v. 24, n. 13, p. 11049, July/2023. Available at: |
| [36] | SOFIA, C. et al. Promising advances in treatments for the management of idiopathic pulmonary fibrosis. Expert Opin Pharmacother, Italy, v. 25, n. 6, p. 717-725, April/2024. Available at: |
| [37] |
SOUZA, R. L. D; FRANCO, L. G. M. D. M; SIQUEIRA, M. C. D. General Approach to Idiopathic Pulmonary Fibrosis, Brazil, v. 23, n. 3, p. 1-7, March/2023. Available at:
https://doi.org/10.25248/REAMed.e12623.2023 (Accessed on: 5 Aug. 2024). |
| [38] | STELLA, G. M. et al. The oncogenic landscape of the idiopathic pulmonary fibrosis: a narrative review. Trans Lung Cancer Res, Italy, v. 11, n. 3, p. 472-496, March. /2022. Available at: |
| [39] |
SYNAPSE - Idiopathic Pulmonary Fibrosis: Current Clinical Research Status Targeting GPCR. Available at:
https://synapse.patsnap.com/blog/idiopathic-pulmonary-fibrosis-current-clinical-research-status-targeting-gpcr (Accessed on: 6 Sept. 2024). |
| [40] | TIRELLI, C. et al. The Genetic and Epigenetic Footprint in Idiopathic Pulmonary Fibrosis and Familial Pulmonary Fibrosis: A State-of-the-Art Review. Diagnostics (Basel), Italy, v. 12, n. 12, p. 3107, Dec./2022) |
| [41] | TOMOS, I. et al. Molecular and Genetic Biomarkers in Idiopathic Pulmonary Fibrosis: Where Are We Now? Biomedicines, Greece v. 11, n. 10, p. 2796, Oct./2023. Available at: |
| [42] | VOLKMANN, E. R. et al. Lysophosphatidic acid receptor 1 inhibition: a potential treatment target for pulmonary fibrosis. Eur Respir Rev, USA, v. 33, n. 172, p. 240015, June/2024. Available at: |
| [43] | YANG, S. et al. Therapeutic Applications of Mesenchymal Stem Cells in Idiopathic Pulmonary Fibrosis. Front Cell Dev Biol, China, v. 9, n. 9, p. 639657, March/2021. Available at: |
| [44] | YANG, Y. et al. Mesenchymal stem cells and pulmonary fibrosis: a bibliometric and visualization analysis of literature published between 2002 and 2021. Frontiers, China, v. 4, n. 1, p. 1, July/2023. Available at: |
| [45] |
YOSHIHARA, M. et al. Administration of Collagen Peptide Prevents the Progression of Pulmonary Fibrosis in Bleomycin-Treated Mice. Biologics, Japan, v. 3, n. 3, p. 187-197, July/2023. Available at:
https://doi.org/10.3390/biologics3030010 (Accessed on: 6 Jan. 2025). |
APA Style
Ferreira, J. S., Mito, G., Hermenegildo, E. (2025). Regenerative Therapies in the Treatment of Idiopathic Pulmonary Fibrosis: A Literature Review. American Journal of Clinical and Experimental Medicine, 13(4), 99-134. https://doi.org/10.11648/j.ajcem.20251304.15
ACS Style
Ferreira, J. S.; Mito, G.; Hermenegildo, E. Regenerative Therapies in the Treatment of Idiopathic Pulmonary Fibrosis: A Literature Review. Am. J. Clin. Exp. Med. 2025, 13(4), 99-134. doi: 10.11648/j.ajcem.20251304.15
@article{10.11648/j.ajcem.20251304.15,
author = {Janislene S Ferreira and Gislaine Mito and Elaine Hermenegildo},
title = {Regenerative Therapies in the Treatment of Idiopathic Pulmonary Fibrosis: A Literature Review
},
journal = {American Journal of Clinical and Experimental Medicine},
volume = {13},
number = {4},
pages = {99-134},
doi = {10.11648/j.ajcem.20251304.15},
url = {https://doi.org/10.11648/j.ajcem.20251304.15},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajcem.20251304.15},
abstract = {Idiopathic Pulmonary Fibrosis (IPF) is a disease that leads to respiratory failure and subsequent death. To prevent chronic lung diseases from rising further up the list of leading causes of death worldwide, new innovative therapeutic approaches are needed. This work aims to present a literature review on advances in regenerative therapies in the treatment of IPF. The bibliographic strategy of this research used papers published in last ten years taken from the electronic databases PubMed and Capes Periodical Portal, which include the current understanding of this pathology, and the different treatment modalities. The majority of selected articles were concentrated in the last three years of this review, corroborating the expectation of recent advancements given the current massive scientific research on IPF. This review focused on current treatments, their limitations and investigated cutting-edge research in regenerative therapies. The research resulted in the presentation of ongoing studies and therapies segregated between pharmacological manipulation and the use of stem cells. Both categories of treatment focus on restoring endogenous lung repair or targeting pathways that inhibit dysregulated regeneration. Genetic and epigenetic factors were constantly highlighted as extremely necessary for the diagnosis. Conclusively, given the heterogeneity of this pathology, the authors propose, for the next general IPF treatment protocol, the inclusion of a combination of therapies, accentuating the pro and post-installed disease components. Encouraging the established of specialized health centers is also part of the findings of this research. They promote close cooperation between pulmonologists, radiologists, biomedical geneticists, pathologists, among others.},
year = {2025}
}
TY - JOUR T1 - Regenerative Therapies in the Treatment of Idiopathic Pulmonary Fibrosis: A Literature Review AU - Janislene S Ferreira AU - Gislaine Mito AU - Elaine Hermenegildo Y1 - 2025/08/18 PY - 2025 N1 - https://doi.org/10.11648/j.ajcem.20251304.15 DO - 10.11648/j.ajcem.20251304.15 T2 - American Journal of Clinical and Experimental Medicine JF - American Journal of Clinical and Experimental Medicine JO - American Journal of Clinical and Experimental Medicine SP - 99 EP - 134 PB - Science Publishing Group SN - 2330-8133 UR - https://doi.org/10.11648/j.ajcem.20251304.15 AB - Idiopathic Pulmonary Fibrosis (IPF) is a disease that leads to respiratory failure and subsequent death. To prevent chronic lung diseases from rising further up the list of leading causes of death worldwide, new innovative therapeutic approaches are needed. This work aims to present a literature review on advances in regenerative therapies in the treatment of IPF. The bibliographic strategy of this research used papers published in last ten years taken from the electronic databases PubMed and Capes Periodical Portal, which include the current understanding of this pathology, and the different treatment modalities. The majority of selected articles were concentrated in the last three years of this review, corroborating the expectation of recent advancements given the current massive scientific research on IPF. This review focused on current treatments, their limitations and investigated cutting-edge research in regenerative therapies. The research resulted in the presentation of ongoing studies and therapies segregated between pharmacological manipulation and the use of stem cells. Both categories of treatment focus on restoring endogenous lung repair or targeting pathways that inhibit dysregulated regeneration. Genetic and epigenetic factors were constantly highlighted as extremely necessary for the diagnosis. Conclusively, given the heterogeneity of this pathology, the authors propose, for the next general IPF treatment protocol, the inclusion of a combination of therapies, accentuating the pro and post-installed disease components. Encouraging the established of specialized health centers is also part of the findings of this research. They promote close cooperation between pulmonologists, radiologists, biomedical geneticists, pathologists, among others. VL - 13 IS - 4 ER -
The Division of Biomedical Science, School of Biomedicine, Estacio De Sa University, Rio De Janeiro, Brazil
The Division of Biomedical Science, School of Biomedicine, Estacio De Sa University, Rio De Janeiro, Brazil
The Division of Biomedical Science, School of Biomedicine, Estacio De Sa University, Rio De Janeiro, Brazil
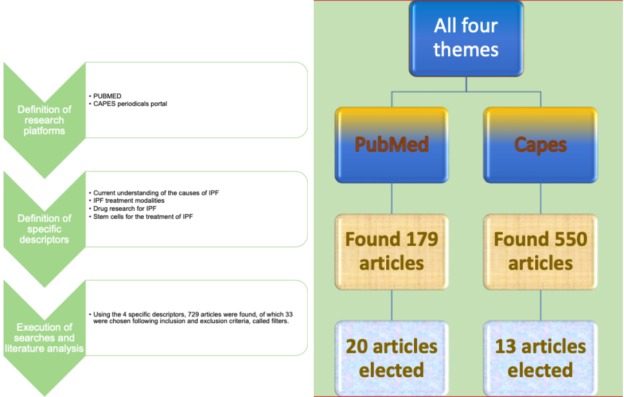
Figure 1. Flowchart demonstrating the research steps and the result.
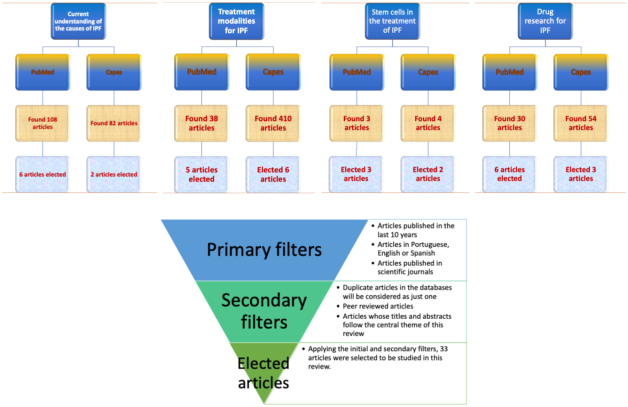
Figure 2. Flowchart demonstrating the research steps for the four specific descriptors in the electronic databases, as well as the inclusion and exclusion criteria called filters.
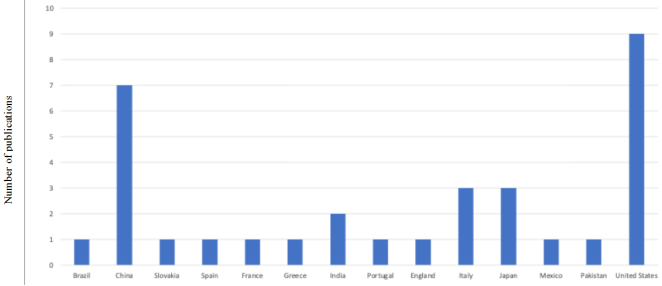
Figure 3. Bibliometric analysis of countries and number of publications used.
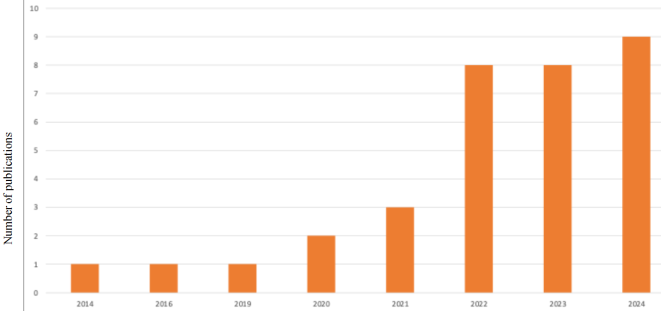
Figure 4. Bibliometric analysis of the year and number of publications used.
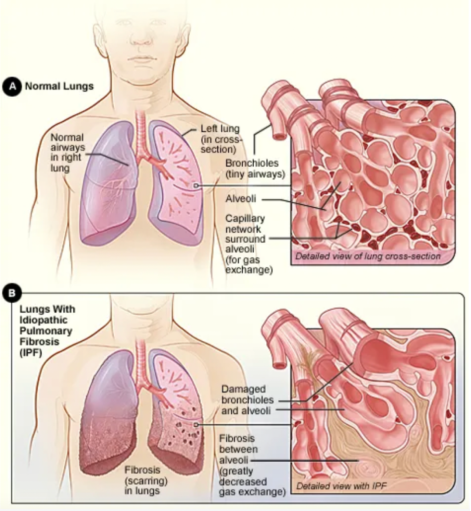
Figure 5. Comparison between a healthy lung (A) and a diseased lung (B).
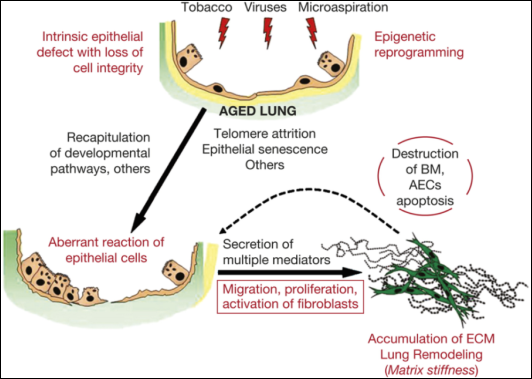
Figure 6. Integral model involved in the pathogenesis of IPF.
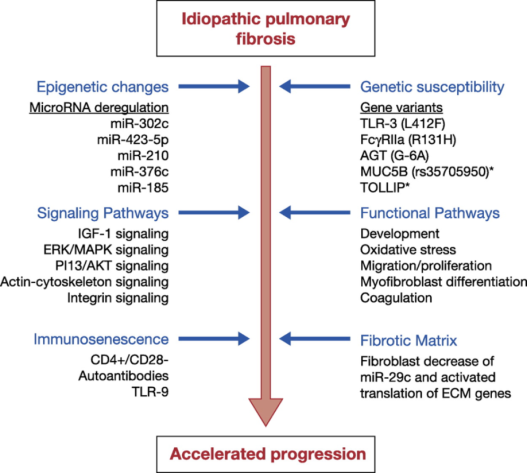
Figure 7. Mechanisms associated with the acceleration rate of IPF.
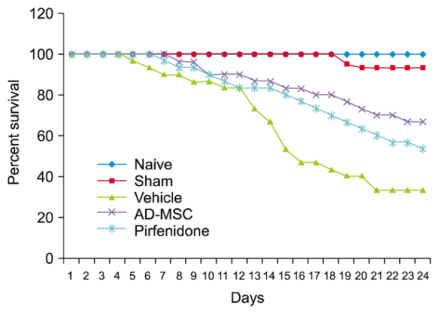
Figure 8. Survival estimates for animals with bleomycin-induced pulmonary fibrosis.
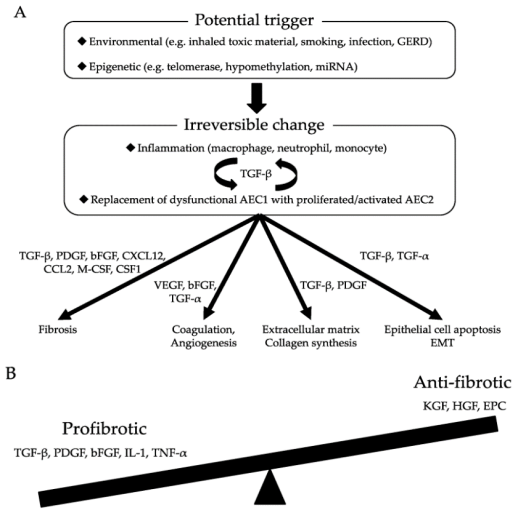
Figure 9. (A) Molecular mechanisms of pulmonary fibrosis; (B) Imbalance of pro-fibrotic and anti-fibrotic mediators leads to defective regeneration and aberrant remodeling, resulting in the pathological transformation of pulmonary fibrosis.
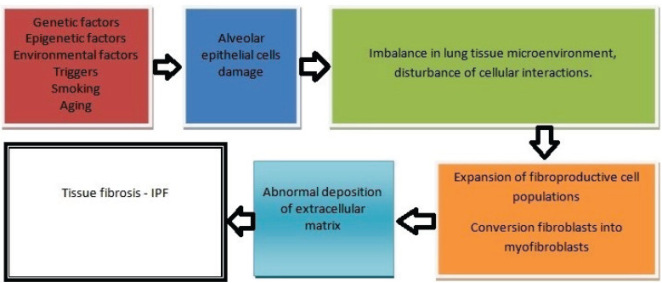
Figure 10. Pathogenesis of IPF.
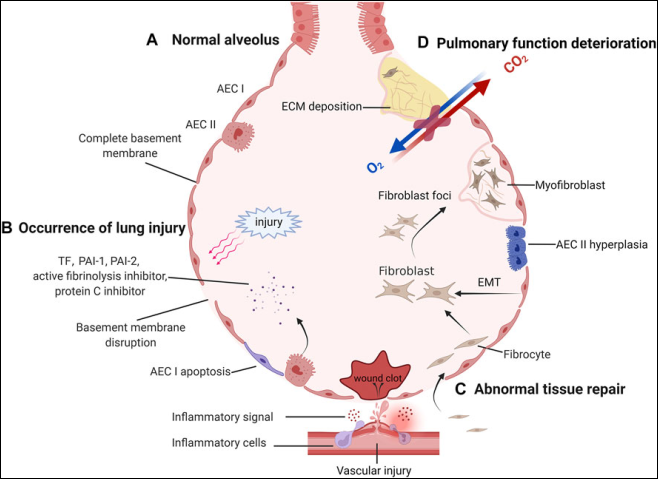
Figure 11. Pathological process of IPF.
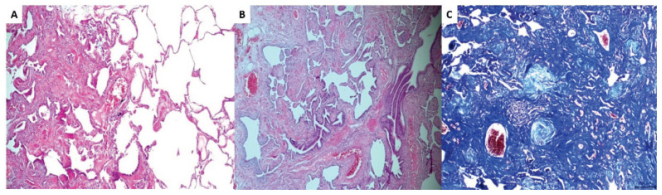
Figure 12. Histological pattern compatible with IPF.
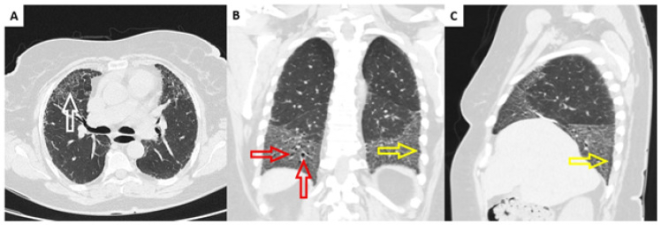
Figure 13. Computed tomography of the lungs in a patient diagnosed with IPF.
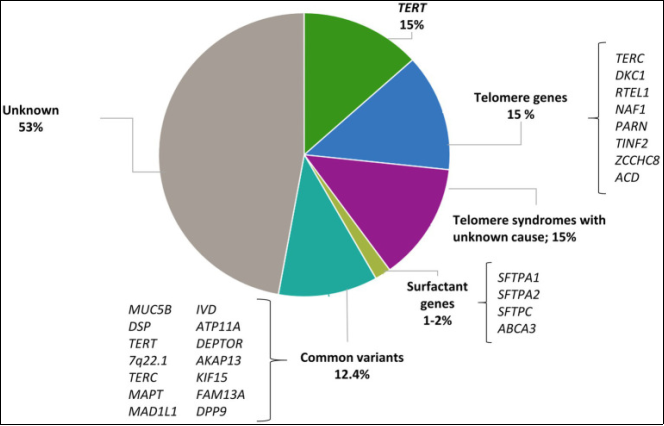
Figure 14. Contribution of rare and common variants to IPF risk.
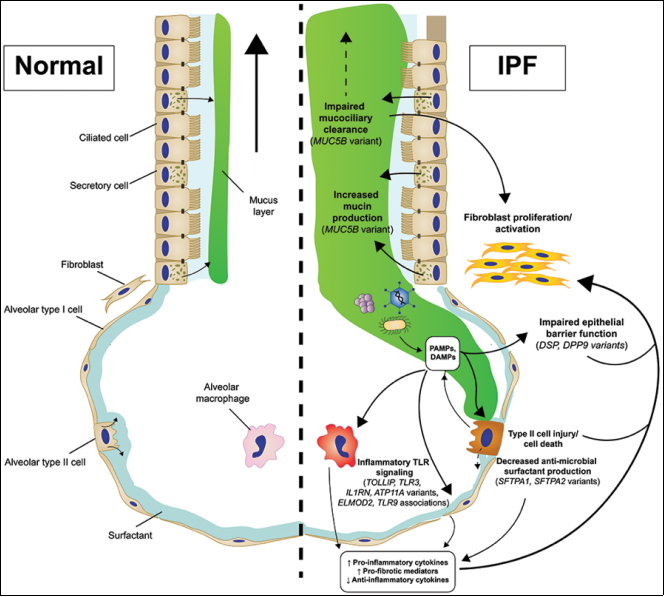
Figure 15. Changes in innate immunity of genetic origin contributing to the pathogenesis of IPF.
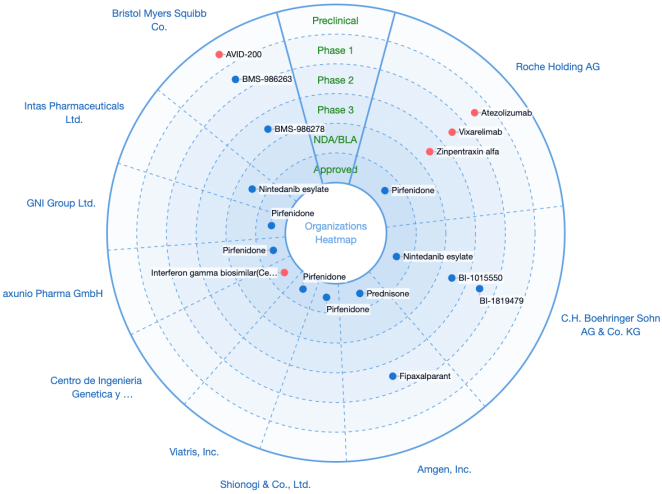
Figure 16. Global statistics on drug research (chemicals and biologicals) in IPF.
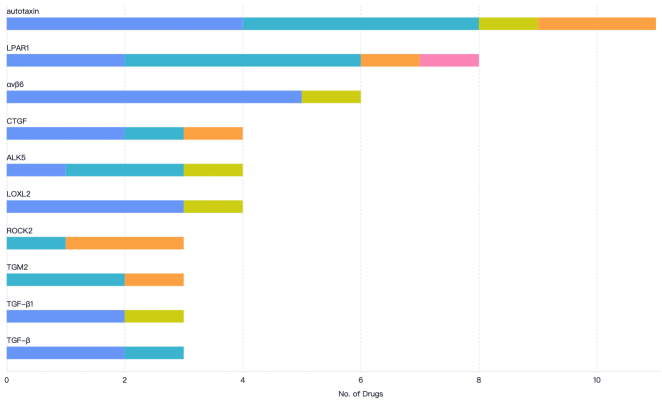
Figure 17. The ten most popular targets investigated for the treatment of IPF.
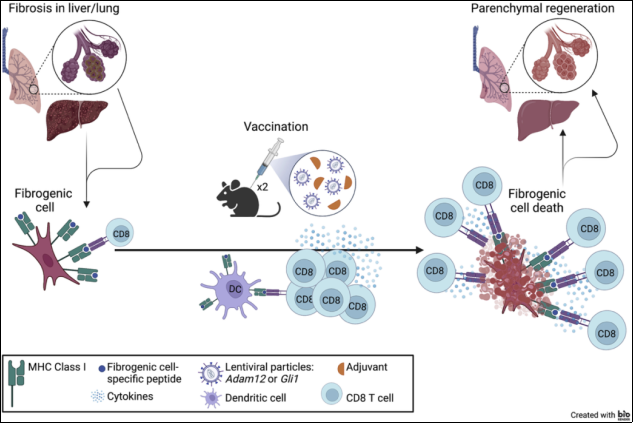
Figure 18. Scheme of immunotherapeutic vaccination.
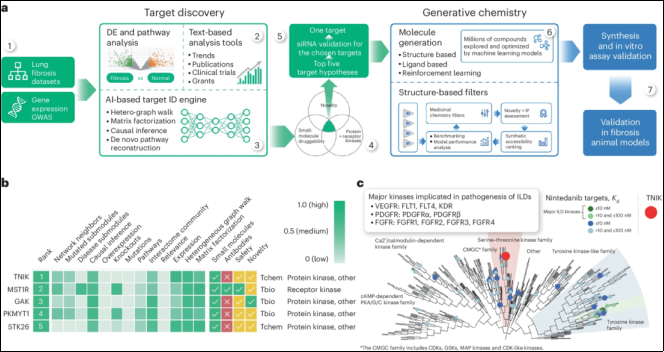
Figure 19. Predictive Artificial Intelligence (AI) approach to identify antifibrotic target.
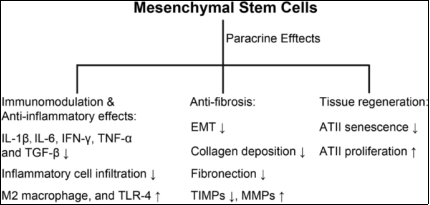
Figure 20. Mechanism of MSC-based therapy for IPF (MSC = mesenchymal stem cells).
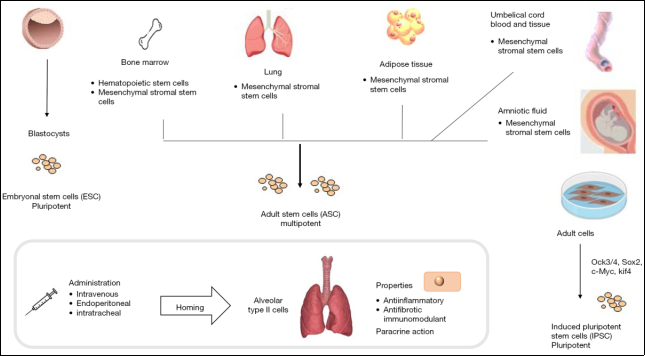
Figure 21. Different types of stem cells for the treatment of IPF.
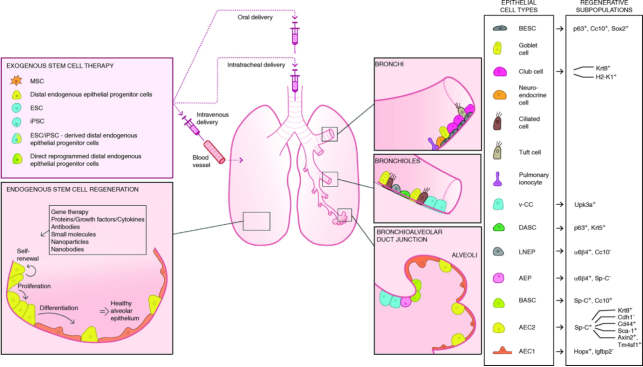
Figure 22. Strategies to promote therapeutic lung regeneration in IPF.
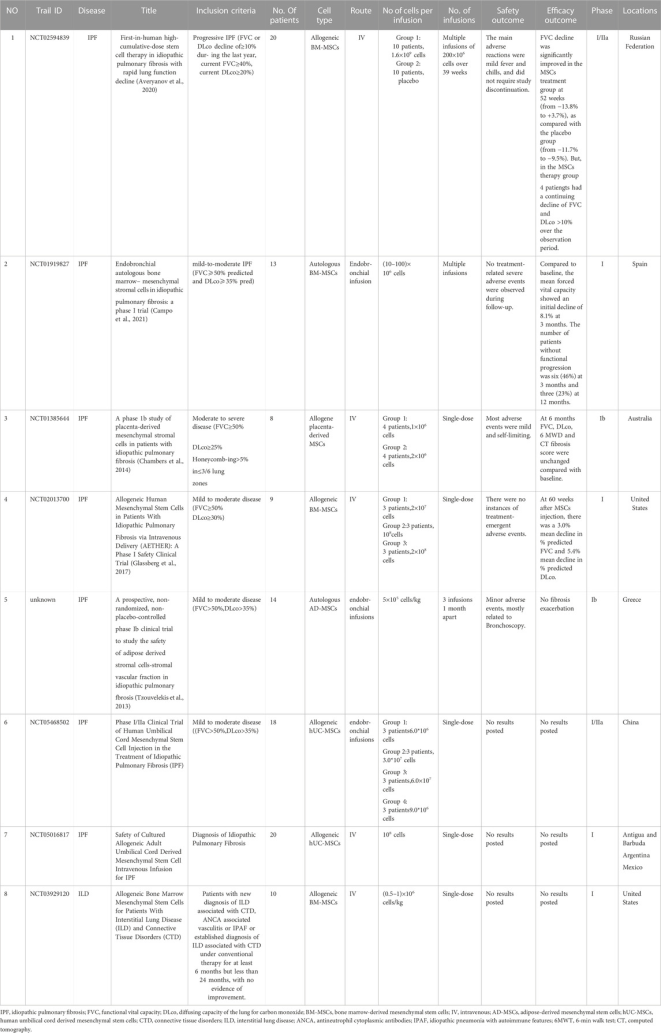
Figure 23. Summary of current clinical trials on MSCs and MSC-EVs (Extracellular Vesicles) for IPF.
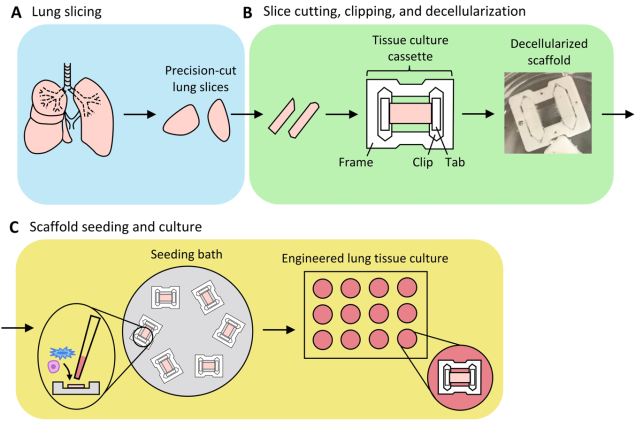
Figure 24. Schematic of the preparation of engineered lung tissue.
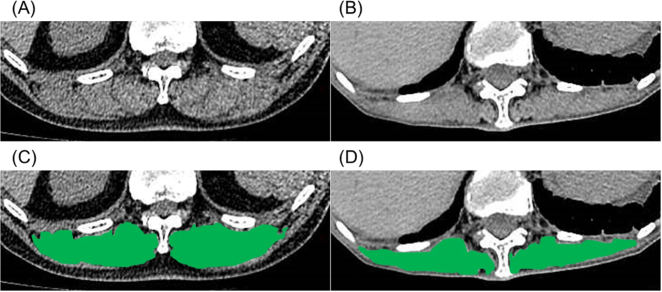
Figure 25. Representative computed tomography images used to measure the cross-sectional area of the erector spinae muscles (A, B).
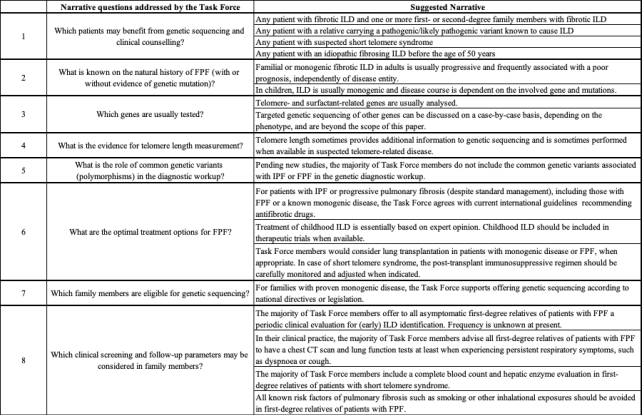
Figure 26. Issues, by consensus, addressed by the task force and their respective suggestive narrative.
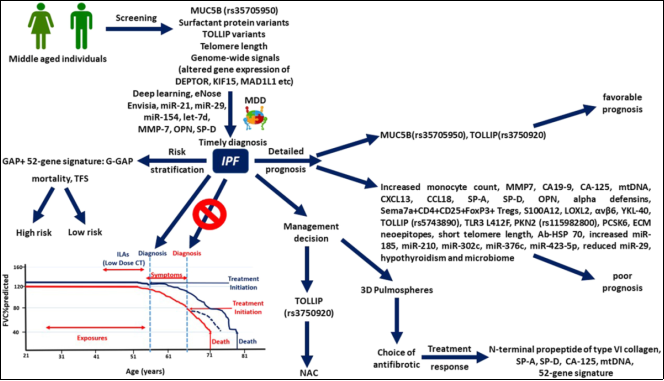
Figure 27. Schematic representation of personalized medicine approaches for patients with IPF.
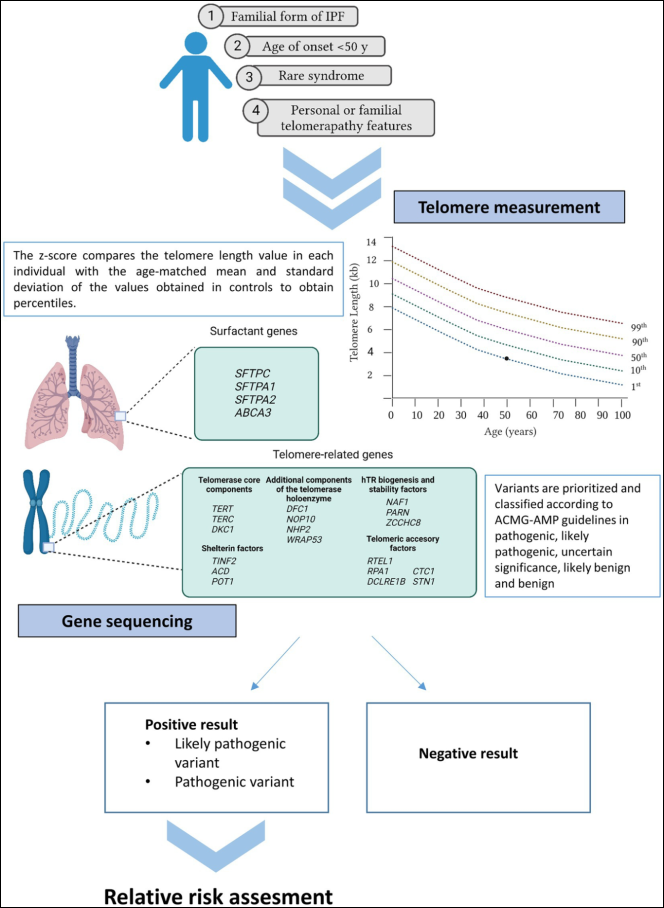
Figure 28. Current recommendations for genetic testing in IPF.
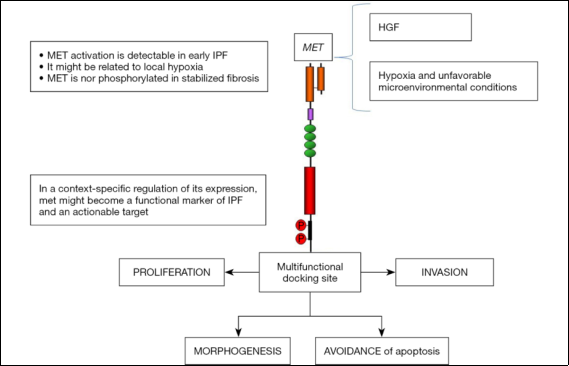
Figure 29. MET oncogene factor as an actionable target for IPF.
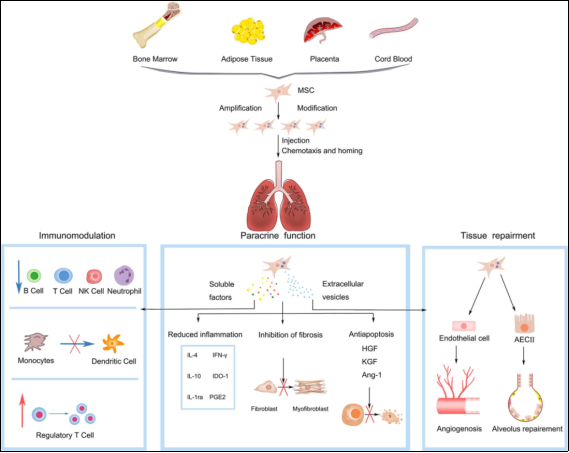
Figure 30. Summary of therapeutic properties and mechanisms of mesenchymal stem cells in IPF.
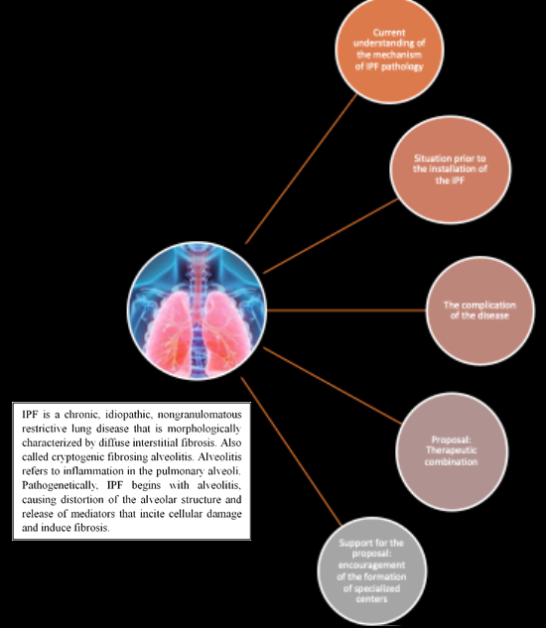
Figure 31. Visual representation of a summary of this research.

Figure 32. Visual representation of a summary of mechanisms, complicating factors, and therapeutic combinations for IPF.
Information

