Alzheimer’s disease (AD) is a neurodegenerative condition characterised by several markers and physiological manifestations. While Alzheimer’s disease affects millions of people throughout the world, the intricacy of the condition and the limits of experimental models have slowed the discovery of viable treatments. Rodent models helped researchers identify critical features of Alzheimer’s disease pathogenesis and test novel treatment strategies. This chapter gives a detailed summary of rodent models used in Alzheimer’s disease research, concentrating on the numerous types of transgenic, knock-in and knock-out models that replicate the genetic alterations linked with familial AD. We look into pharmacological and neurotoxin-induced models as well as infusion models, to imitate particular pathological characteristics of the disease. In these models, pathological assessments are essential for determining the development of amyloid plaque, hyperphosphorylation of tau and neuroinflammatory responses, immunohistochemistry, ELISA as well as synaptic marker investigations, all play significant contributions. Regardless of the benefits they provide, rodent models have substantial limitations in recreating the complete spectrum of human Alzheimer’s disease, notable the neurodegeneration and comorbidities present in sporadic AD. As a result, the research is shifting toward more advanced humanised models and gene-editing tools, such as CRISPR/Cas9, to eliminate the disparity between research on rodents and human therapeutic applications. This chapter finishes with a discussion of future AD research paths, highlighting the importance of improved models that combine environmental, genetic, and lifestyle components in order to better portray the complexity of AD. Rodent models are still in an angle to contribute significantly to our understanding of AD and the development of disease-transforming treatments by overcoming these constraints.
| Published in | Science Discovery Medicine (Volume 1, Issue 2) |
| DOI | 10.11648/j.sdmed.20260102.14 |
| Page(s) | 85-98 |
| Creative Commons |
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited. |
| Copyright |
Copyright © The Author(s), 2026. Published by Science Publishing Group |
Alzheimer’s Disease, Rodent Models, Amyloid Plaques, Tau Hyperphosphorylation
Tg2576 | APPswe (Swedish Mutation) | Amyloid plaques, synaptic loss | Memory deficits (Morris Water Maze, Novel Object Recognition) | Early-stage amyloid pathology, synaptic dysfunction | [25] |
PDAPP | APP (Indiana Mutation) | Early amyloid deposition, neuroinflammation | Severe cognitive impairments | Role of amyloid in neurodegeneration | [26] |
3xTg-AD | APP (Swedish), PSEN1, Tau | Amyloid plaques, tau tangles, neurodegeneration | Memory impairments, deficits in spatial learning | Interaction between amyloid and tau pathologies | [27] |
5xFAD | APP (Swedish, Florida), PSEN1 | Rapid amyloid plaque deposition, synaptic dysfunction | Cognitive decline, LTP/LTD impairments | Early-onset model, role of Aβ in synaptic toxicity and memory loss | [28] |
Tau P310L | Tau (P310L mutation) | Neurofibrillary tangles, neuronal loss | Motor and cognitive impairments | Role of tau in neurodegeneration and cognitive deficits | [29] |
Aβ | Amyloid-beta |
AD | Alzheimer’s Disease |
AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid |
APP | Amyloid Precursor Protein |
CNS | Central Nervous System |
CRISPR | Clustered Regularly Interspaced Short Palin-dromic Repeats |
dCas9 | Deactivated Form of Cas9 |
ELISA | Enzyme-linked Immunosorbent Assay |
FAD | Familial Alzheimer’s Disease |
GFAP | Glial Fibrillary Acidic Protein |
gRNAs | Guide RNAs |
GWAS | Genome-wide Association Studies |
Iba1 | Ionized Calcium-binding Adaptor Molecule 1 |
IHC | Immunohistochemistry |
IL-1 | Interleukin-1 |
IL-6 | Interleukin-6 |
LTD | Long-term Depression |
LTP | Long-term Potentiation |
MWM | Morris Water Maze |
NFTs | Neurofibrillary Tangles |
NMDA | N-methyl-D-aspartate |
NOR | Novel Object Recognition Test |
PHFs | Paired Helical Filaments |
PSD95 | Postsynaptic Density Protein 95 |
PSEN1 | Presenilin-1 |
PSEN2 | Presenilin-2 |
SNPs | Single Nucleotide Polymorphisms |
TALENs | Transcription Activator-like Effector Nucleases |
TNF | Tumor Necrosis Factor |
ZFN | Zinc Finger Nuclease |
| [1] | J. Götz, L. G. Bodea, and M. Goedert, ‘Rodent models for Alzheimer disease’, Oct. 01, 2018, Nature Publishing Group. |
| [2] | P. Sethi et al., ‘Exploring advancements in early detection of Alzheimer’s disease with molecular assays and animal models’, Sep. 01, 2024, Elsevier Ireland Ltd. |
| [3] | K. J. Egan, H. M. Vesterinen, V. Beglopoulos, E. S. Sena, and M. R. Macleod, ‘From a mouse: systematic analysis reveals limitations of experiments testing interventions in Alzheimer’s disease mouse models’, Evid. Based. Preclin. Med., vol. 3, no. 1, pp. 12-23, Aug. 2016, |
| [4] | N. Braidy et al., ‘Recent rodent models for Alzheimer’s disease: Clinical implications and basic research’, Feb. 2012. |
| [5] | M. Newman, E. Ebrahimie, and M. Lardelli, ‘Using the zebrafish model for Alzheimer’s disease research’, 2014, Frontiers Research Foundation. |
| [6] | S. J. Sukoff Rizzo and J. N. Crawley, ‘Behavioral Phenotyping Assays for Genetic Mouse Models of Neurodevelopmental, Neurodegenerative, and Psychiatric Disorders’, Feb. 08, 2017, Annual Reviews Inc. |
| [7] | C. Li and J. Götz, ‘Tau-based therapies in neurodegeneration: opportunities and challenges’, Nat. Rev. Drug Discov., vol. 16, no. 12, pp. 863-883, Dec. 2017, |
| [8] | P. Scheltens et al., ‘Alzheimer’s disease’, The Lancet, vol. 397, no. 10284, pp. 1577-1590, Apr. 2021, |
| [9] | N. Weishaupt et al., ‘APP21 transgenic rats develop age-dependent cognitive impairment and microglia accumulation within white matter tracts’, J. Neuroinflammation, vol. 15, no. 1, p. 241, Dec. 2018, |
| [10] | L. M. De Plano, G. Calabrese, S. Conoci, S. P. P. Guglielmino, S. Oddo, and A. Caccamo, ‘Applications of CRISPR-Cas9 in Alzheimer’s Disease and Related Disorders’, Aug. 01, 2022, MDPI. |
| [11] | A. Doyle, M. P. McGarry, N. A. Lee, and J. J. Lee, ‘The construction of transgenic and gene knockout/knockin mouse models of human disease’, Transgenic Res., vol. 21, no. 2, pp. 327-349, Apr. 2012, |
| [12] | L. D’Adamio, ‘Transfixed by transgenics: how pathology assumptions are slowing progress in Alzheimer’s disease and related dementia research’, EMBO Mol. Med., vol. 15, no. 11, Nov. 2023, |
| [13] | T. Saito et al., ‘Single App knock-in mouse models of Alzheimer’s disease’, Nat. Neurosci., vol. 17, no. 5, pp. 661-663, May 2014, |
| [14] | S. Khan, K. H. Barve, and M. S. Kumar, ‘Recent Advancements in Pathogenesis, Diagnostics and Treatment of Alzheimer’s Disease’, Curr. Neuropharmacol., vol. 18, no. 11, pp. 1106-1125, Nov. 2020, |
| [15] | L. Jia et al., ‘Concordance between the assessment of Aβ42, T‐tau, and P‐T181‐tau in peripheral blood neuronal‐derived exosomes and cerebrospinal fluid’, Alzheimer’s & Dementia, vol. 15, no. 8, pp. 1071-1080, Aug. 2019, |
| [16] | Z. Breijyeh and R. Karaman, ‘Comprehensive Review on Alzheimer’s Disease: Causes and Treatment’, Molecules, vol. 25, no. 24, p. 5789, Dec. 2020, |
| [17] | S.-E. Lee et al., ‘Production of transgenic pig as an Alzheimer’s disease model using a multi-cistronic vector system’, PLoS One, vol. 12, no. 6, p. e0177933, Jun. 2017, |
| [18] | J. J. Sabbagh, J. W. Kinney, and J. L. Cummings, ‘Animal systems in the development of treatments for Alzheimer’s disease: challenges, methods, and implications’, Neurobiol. Aging, vol. 34, no. 1, pp. 169-183, Jan. 2013, |
| [19] | L. Tesson et al., ‘Transgenic Modifications of the Rat Genome’, Transgenic Res., vol. 14, no. 5, pp. 531-546, Oct. 2005, |
| [20] | J. C. Polanco, C. Li, L.-G. Bodea, R. Martinez-Marmol, F. A. Meunier, and J. Götz, ‘Amyloid-β and tau complexity — towards improved biomarkers and targeted therapies’, Nat. Rev. Neurol., vol. 14, no. 1, pp. 22-39, Jan. 2018, |
| [21] | M. Kitazawa, R. Medeiros, and F. M. LaFerla, ‘Transgenic Mouse Models of Alzheimer Disease: Developing a Better Model as a Tool for Therapeutic Interventions’, Curr. Pharm. Des., vol. 18, no. 8, pp. 1131-1147, Mar. 2012, |
| [22] | R. Cacace, K. Sleegers, and C. Van Broeckhoven, ‘Molecular genetics of early‐onset Alzheimer’s disease revisited’, Alzheimer’s & Dementia, vol. 12, no. 6, pp. 733-748, Jun. 2016, |
| [23] | R. Belfiore et al., ‘Temporal and regional progression of Alzheimer’s disease‐like pathology in 3xTg‐AD mice’, Aging Cell, vol. 18, no. 1, Feb. 2019, |
| [24] | J. Coomaraswamy et al., ‘Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease’, Proceedings of the National Academy of Sciences, vol. 107, no. 17, pp. 7969-7974, Apr. 2010, |
| [25] | C. Haass et al., ‘The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway’, Nat. Med., vol. 1, no. 12, pp. 1291-1296, Dec. 1995, |
| [26] | C. Bi, S. Bi, and B. Li, ‘Processing of Mutant β-Amyloid Precursor Protein and the Clinicopathological Features of Familial Alzheimer’s Disease’, Aging Dis., vol. 10, no. 2, p. 383, 2019, |
| [27] | A. R. Roda, G. Esquerda-Canals, J. Martí-Clúa, and S. Villegas, ‘Cognitive Impairment in the 3xTg-AD Mouse Model of Alzheimer’s Disease is Affected by Aβ-ImmunoTherapy and Cognitive Stimulation’, Pharmaceutics, vol. 12, no. 10, p. 944, Oct. 2020, |
| [28] | A. L. Oblak et al., ‘Comprehensive Evaluation of the 5XFAD Mouse Model for Preclinical Testing Applications: A MODEL-AD Study’, Front. Aging Neurosci., vol. 13, Jul. 2021, |
| [29] | M. C. Silva et al., ‘Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models’, Elife, vol. 8, Mar. 2019, |
| [30] | M. Noetzli and C. B. Eap, ‘Pharmacodynamic, Pharmacokinetic and Pharmacogenetic Aspects of Drugs Used in the Treatment of Alzheimer’s Disease’, Clin. Pharmacokinet., vol. 52, no. 4, pp. 225-241, Apr. 2013, |
| [31] | C. H. Poon, Y. Wang, M. L. Fung, C. Zhang, and L. W. Lim, ‘Rodent models of amyloid-beta feature of alzheimer’s disease: Development and potential treatment implications’, Oct. 01, 2020, International Society on Aging and Disease. |
| [32] | S. E. Marsh et al., ‘HuCNS-SC Human NSCs Fail to Differentiate, Form Ectopic Clusters, and Provide No Cognitive Benefits in a Transgenic Model of Alzheimer’s Disease’, Stem Cell Reports, vol. 8, no. 2, pp. 235-248, Feb. 2017, |
| [33] | J. Cummings, G. Lee, A. Ritter, M. Sabbagh, and K. Zhong, ‘Alzheimer’s disease drug development pipeline: 2019’, Alzheimer’s & Dementia: Translational Research & Clinical Interventions, vol. 5, no. 1, pp. 272-293, Jan. 2019, |
| [34] | P. K. Dash et al., ‘Humanized Mice for Infectious and Neurodegenerative disorders’, Dec. 01, 2021, BioMed Central Ltd. |
| [35] | C. Salazar, G. Valdivia, álvaro O. Ardiles, J. Ewer, and A. G. Palacios, ‘Genetic variants associated with neurodegenerative Alzheimer disease in natural models’, Jan. 06, 2016, BioMed Central Ltd. |
| [36] | J. Verheijen and K. Sleegers, ‘Understanding Alzheimer Disease at the Interface between Genetics and Transcriptomics’, Trends in Genetics, vol. 34, no. 6, pp. 434-447, Jun. 2018, |
| [37] | L. Gao, Y. Zhang, K. Sterling, and W. Song, ‘Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential’, Transl. Neurodegener., vol. 11, no. 1, p. 4, Jan. 2022, |
| [38] | D. Van Dam and P. P. De Deyn, ‘Non human primate models for Alzheimer’s disease-related research and drug discovery’, Expert Opin. Drug Discov., vol. 12, no. 2, pp. 187-200, Feb. 2017, |
| [39] | A. Nazem and G. A. Mansoori, ‘Nanotechnology Solutions for Alzheimer’s Disease: Advances in Research Tools, Diagnostic Methods and Therapeutic Agents’, Journal of Alzheimer’s Disease, vol. 13, no. 2, pp. 199-223, Mar. 2008, |
| [40] | B. C. Gonçalves et al., ‘Antiviral therapies: advances and perspectives’, Fundam. Clin. Pharmacol., vol. 35, no. 2, pp. 305-320, Apr. 2021, |
| [41] | G. Esquerda-Canals, L. Montoliu-Gaya, J. Güell-Bosch, and S. Villegas, ‘Mouse Models of Alzheimer’s Disease’, Journal of Alzheimer’s Disease, vol. 57, no. 4, pp. 1171-1183, Apr. 2017, |
| [42] | W. M. Song, S. Joshita, Y. Zhou, T. K. Ulland, S. Gilfillan, and M. Colonna, ‘Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism’, Journal of Experimental Medicine, vol. 215, no. 3, pp. 745-760, Mar. 2018, |
| [43] | S. Journal and U. Journal, ‘International Journal of Recent Technology and Engineering (IJRTE)’, 2020, |
| [44] | J. A. Trejo-Lopez, A. T. Yachnis, and S. Prokop, ‘Neuropathology of Alzheimer’s Disease’, Neurotherapeutics, vol. 19, no. 1, pp. 173-185, Jan. 2022, |
| [45] | P. Sethi et al., ‘Exploring advancements in early detection of Alzheimer’s disease with molecular assays and animal models’, Ageing Res. Rev., vol. 100, p. 102411, Sep. 2024. |
| [46] | C. Agca et al., ‘Development of transgenic rats producing human β-amyloid precursor protein as a model for Alzheimer’s disease: Transgene and endogenous APP genes are regulated tissue-specifically’, BMC Neurosci., vol. 9, no. 1, p. 28, Dec. 2008, |
| [47] | S. L. Espíndola et al., ‘Modulation of Tau Isoforms Imbalance Precludes Tau Pathology and Cognitive Decline in a Mouse Model of Tauopathy’, Cell Rep., vol. 23, no. 3, pp. 709-715, Apr. 2018, |
| [48] | F. Clavaguera et al., ‘Transmission and spreading of tauopathy in transgenic mouse brain’, Nat. Cell Biol., vol. 11, no. 7, pp. 909-913, Jul. 2009, |
| [49] | T. A. Pascoal et al., ‘Amyloid-β and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer’s disease’, Mol. Psychiatry, vol. 22, no. 2, pp. 306-311, Feb. 2017, |
| [50] | A. Nazem, R. Sankowski, M. Bacher, and Y. Al-Abed, ‘Rodent models of neuroinflammation for Alzheimer’s disease’, Apr. 17, 2015, BioMed Central Ltd. |
| [51] | E. E. Spangenberg et al., ‘Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology’, Brain, vol. 139, no. 4, pp. 1265-1281, Apr. 2016, |
| [52] | J. Beauquis et al., ‘Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease’, Exp. Neurol., vol. 239, pp. 28-37, Jan. 2013, |
| [53] | S. Commins and B. P. Kirby, ‘The complexities of behavioural assessment in neurodegenerative disorders: A focus on Alzheimer’s disease’, Sep. 01, 2019, Academic Press. |
| [54] | K. E. Ameen‐Ali, S. B. Wharton, J. E. Simpson, P. R. Heath, P. Sharp, and J. Berwick, ‘Review: Neuropathology and behavioural features of transgenic murine models of Alzheimer’s disease’, Neuropathol. Appl. Neurobiol., vol. 43, no. 7, pp. 553-570, Dec. 2017, |
| [55] | D. L. King and G. W. Arendash, ‘Behavioral characterization of the Tg2576 transgenic model of Alzheimer’s disease through 19 months’, Physiol. Behav., vol. 75, no. 5, pp. 627-642, Apr. 2002, |
| [56] | I. Heggland, I. S. Storkaas, H. T. Soligard, A. Kobro‐Flatmoen, and M. P. Witter, ‘Stereological estimation of neuron number and plaque load in the hippocampal region of a transgenic rat model of Alzheimer’s disease’, European Journal of Neuroscience, vol. 41, no. 9, pp. 1245-1262, May 2015, |
| [57] | F. Kosel, P. Torres Munoz, J. R. Yang, A. A. Wong, and T. B. Franklin, ‘Age-related changes in social behaviours in the 5xFAD mouse model of Alzheimer’s disease’, Behavioural Brain Research, vol. 362, pp. 160-172, Apr. 2019, |
| [58] | E. A. Guzmán et al., ‘Abundance of Aβ5-xlike immunoreactivity in transgenic 5XFAD, APP/PS1KI and 3xTG mice, sporadic and familial Alzheimer’s disease’, Mol. Neurodegener., vol. 9, no. 1, p. 13, Dec. 2014, |
| [59] | P. Filipcik et al., ‘First transgenic rat model developing progressive cortical neurofibrillary tangles’, Neurobiol. Aging, vol. 33, no. 7, pp. 1448-1456, Jul. 2012, |
| [60] | M. J. Winton et al., ‘Intraneuronal APP, Not Free Aβ Peptides in 3xTg-AD Mice: Implications for Tau versus Aβ-Mediated Alzheimer Neurodegeneration’, The Journal of Neuroscience, vol. 31, no. 21, pp. 7691-7699, May 2011, |
| [61] | N. C. Inestrosa et al., ‘Age Progression of Neuropathological Markers in the Brain of the Chilean Rodent Octodon degus, a Natural Model of Alzheimer’s Disease’, Brain Pathology, vol. 25, no. 6, pp. 679-691, Nov. 2015, |
| [62] | F. Stella, J. Laks, J. S. Govone, K. de Medeiros, and O. V. Forlenza, ‘Association of neuropsychiatric syndromes with global clinical deterioration in Alzheimer’s disease patients’, Int. Psychogeriatr., vol. 28, no. 5, pp. 779-786, May 2016, |
APA Style
Singh, A., Duerner, L. (2026). Advances and Limitations of Rodent Models in Alzheimer’s Disease Pathogenesis and Therapeutics. Science Discovery Medicine, 1(2), 85-98. https://doi.org/10.11648/j.sdmed.20260102.14
ACS Style
Singh, A.; Duerner, L. Advances and Limitations of Rodent Models in Alzheimer’s Disease Pathogenesis and Therapeutics. Sci. Discov. Med. 2026, 1(2), 85-98. doi: 10.11648/j.sdmed.20260102.14
@article{10.11648/j.sdmed.20260102.14,
author = {Abhinav Singh and Lena Duerner},
title = {Advances and Limitations of Rodent Models in Alzheimer’s Disease Pathogenesis and Therapeutics},
journal = {Science Discovery Medicine},
volume = {1},
number = {2},
pages = {85-98},
doi = {10.11648/j.sdmed.20260102.14},
url = {https://doi.org/10.11648/j.sdmed.20260102.14},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.sdmed.20260102.14},
abstract = {Alzheimer’s disease (AD) is a neurodegenerative condition characterised by several markers and physiological manifestations. While Alzheimer’s disease affects millions of people throughout the world, the intricacy of the condition and the limits of experimental models have slowed the discovery of viable treatments. Rodent models helped researchers identify critical features of Alzheimer’s disease pathogenesis and test novel treatment strategies. This chapter gives a detailed summary of rodent models used in Alzheimer’s disease research, concentrating on the numerous types of transgenic, knock-in and knock-out models that replicate the genetic alterations linked with familial AD. We look into pharmacological and neurotoxin-induced models as well as infusion models, to imitate particular pathological characteristics of the disease. In these models, pathological assessments are essential for determining the development of amyloid plaque, hyperphosphorylation of tau and neuroinflammatory responses, immunohistochemistry, ELISA as well as synaptic marker investigations, all play significant contributions. Regardless of the benefits they provide, rodent models have substantial limitations in recreating the complete spectrum of human Alzheimer’s disease, notable the neurodegeneration and comorbidities present in sporadic AD. As a result, the research is shifting toward more advanced humanised models and gene-editing tools, such as CRISPR/Cas9, to eliminate the disparity between research on rodents and human therapeutic applications. This chapter finishes with a discussion of future AD research paths, highlighting the importance of improved models that combine environmental, genetic, and lifestyle components in order to better portray the complexity of AD. Rodent models are still in an angle to contribute significantly to our understanding of AD and the development of disease-transforming treatments by overcoming these constraints.},
year = {2026}
}
TY - JOUR T1 - Advances and Limitations of Rodent Models in Alzheimer’s Disease Pathogenesis and Therapeutics AU - Abhinav Singh AU - Lena Duerner Y1 - 2026/04/14 PY - 2026 N1 - https://doi.org/10.11648/j.sdmed.20260102.14 DO - 10.11648/j.sdmed.20260102.14 T2 - Science Discovery Medicine JF - Science Discovery Medicine JO - Science Discovery Medicine SP - 85 EP - 98 PB - Science Publishing Group UR - https://doi.org/10.11648/j.sdmed.20260102.14 AB - Alzheimer’s disease (AD) is a neurodegenerative condition characterised by several markers and physiological manifestations. While Alzheimer’s disease affects millions of people throughout the world, the intricacy of the condition and the limits of experimental models have slowed the discovery of viable treatments. Rodent models helped researchers identify critical features of Alzheimer’s disease pathogenesis and test novel treatment strategies. This chapter gives a detailed summary of rodent models used in Alzheimer’s disease research, concentrating on the numerous types of transgenic, knock-in and knock-out models that replicate the genetic alterations linked with familial AD. We look into pharmacological and neurotoxin-induced models as well as infusion models, to imitate particular pathological characteristics of the disease. In these models, pathological assessments are essential for determining the development of amyloid plaque, hyperphosphorylation of tau and neuroinflammatory responses, immunohistochemistry, ELISA as well as synaptic marker investigations, all play significant contributions. Regardless of the benefits they provide, rodent models have substantial limitations in recreating the complete spectrum of human Alzheimer’s disease, notable the neurodegeneration and comorbidities present in sporadic AD. As a result, the research is shifting toward more advanced humanised models and gene-editing tools, such as CRISPR/Cas9, to eliminate the disparity between research on rodents and human therapeutic applications. This chapter finishes with a discussion of future AD research paths, highlighting the importance of improved models that combine environmental, genetic, and lifestyle components in order to better portray the complexity of AD. Rodent models are still in an angle to contribute significantly to our understanding of AD and the development of disease-transforming treatments by overcoming these constraints. VL - 1 IS - 2 ER -
Institute of Clinical Neurobiology, Julius-Maximilians-University, Wuerzburg, Germany
Faculty of Medicine, University of Cologne, Cologne, Germany
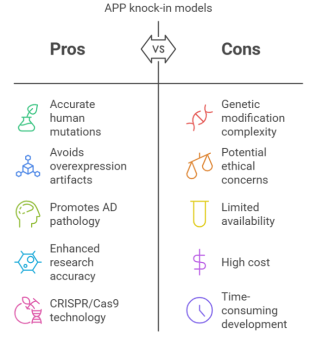
Figure 1. A pictorial representation of pros and cons of APP Knock-in models— The benefits and drawbacks of APP knock-in models in AD research are contrasted in this graphic. These models improve study quality and translatability by precisely stimulating AD pathophysiology, including human-specific mutations and maintaining physiological expression of genes, as well as, avoiding the overexpression artifacts found in conventional transgenic mice while utilizing CRISPR/Cas9 for precise genomic changes.
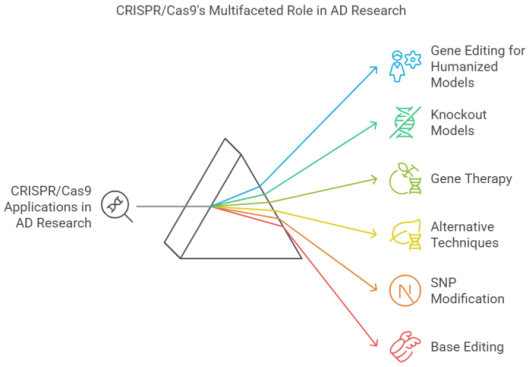
Figure 2. CRISPR/Cas9 Application in AD research – The many uses of CRISPR/Cas9 in AD research is depicted in this figure. Gene editing for humanized models is made possible by CRISPR/Cas9, which introduces genetic variations unique to humans, improving translational accuracy. By specifically deactivating AD-related genes, it makes it easier to crate knockout model as well as used in gene therapy applications to fix mutations linked to the pathology and potentially implement therapeutic treatments. Alternative methods investigate changes such as epigenetic regulations that go beyond traditional gene editing and Base editing offers accurate, permanent nucleotide modifications without double-strand breaks.
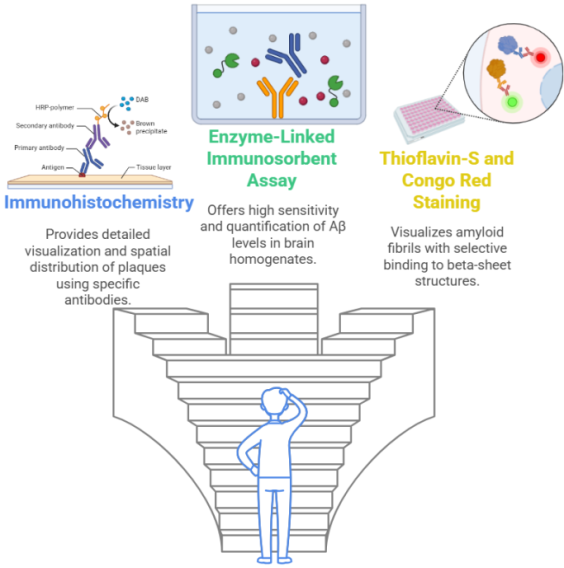
Figure 3. Possible techniques which can be used to assess amyloid plaques in Alzheimer's disease – Three main methods for assessing Aβ plaques in AD research are shown in this illustration. Using particular antibodies, immunohistochemistry helps with histopathological investigation by providing a comprehensive visual representation and geographical distribution of plaques. ELISA is an essential instrument for biochemical evaluation because of its outstanding sensitivity and quantification. The Congo Red enables the detection of aggregates via refraction and fluorescence.
Information

