Pompe disease is a hereditary lysosomal storage disorder characterized by a deficiency in the acid alpha-glucosidase (GAA) enzyme, leading to glycogen accumulation in muscle and neurons. Clinical manifestations vary from severe infantile-onset with hypertrophic cardiomyopathy and early mortality due to respiratory insufficiency to late-onset with proximal muscle weakness, gross motor delay, and progressive respiratory insufficiency. A case of an 11-year-old boy who reported to the pediatric emergency department with a nine-year history of progressive muscle weakness and a one-month history of anemia symptoms (easy fatigue, shortness of breath, pale appearance) and heart failure (orthopnea, dyspnea). His family history included consanguineous marriages and similar conditions in his brother and maternal uncle. On examination, he appeared pale, malnourished, and exhibited signs of respiratory distress and tachypnea. His cardiovascular examination revealed apex beat displacement, elevated JVP, bilateral pedal edema, mild ascites, positive hepatojugular reflux, and systolic murmurs. Respiratory examination indicated bilateral crepitation and wheezes. Musculoskeletal examination showed decreased muscle mass and power, especially in proximal muscles. Abdominal examination revealed hepatosplenomegaly and mild ascites. Radiological findings included an enlarged cardiac shadow with pleural effusion and bilateral radio-opaque shadows on chest x-ray, while echocardiography showed impaired left ventricular systolic function with mild to moderate mitral and tricuspid regurgitation. Laboratory tests indicated elevated aspartate aminotransferase, LDH, and creatine kinase levels, along with normocytic, normochromic anemia. Muscle biopsy from the hamstring revealed PAS stain positive granules. These clinical, radiological, and laboratory findings strongly suggest late-onset Pompe disease, marking this as potentially the second reported case in Pakistan.
This is an Open Access article, distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution and reproduction in any medium or format, provided the original work is properly cited.
Heart Failure, Late-Onset, Pompe's Disease, Case Rare Report, Child
1. Introduction
Pompe disease is an autosomal recessive disorder characterized by the intralysosomal accumulation of glycogen in various body tissues, most notably within the heart and skeletal muscles. This condition is caused by a deficiency of the enzyme acid alpha-glucosidase (GAA; EC 3.2.1.20) due to pathogenic variations in the GAA gene
[1]
Fuller DD, ElMallah MK, Smith BK, et al. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol. 2013; 189(2): 241-249.
Hypertrophic cardiomyopathy and skeletal muscle weakening arise from the accumulation of glycogen in muscle lysosomes. The age of onset and severity of symptoms vary depending on the residual enzymatic activity of GAA. Additionally, the buildup and dysfunction of glycogen in the spinal cord's inner horn and brain neuronal glycogen exacerbate skeletal myopathy
[1]
Fuller DD, ElMallah MK, Smith BK, et al. The respiratory neuromuscular system in Pompe disease. Respir Physiol Neurobiol. 2013; 189(2): 241-249.
Infantile-onset Pompe disease (IOPD) results from a significant deficiency of the lysosomal GAA enzyme, leading to severe hypertrophic cardiomyopathy, marked hypotonia, respiratory failure, and heart failure within the first two months of life. If left undiagnosed, individuals typically succumb to cardiopulmonary complications within the first fourteen months
[2]
Wang RY. A Newborn Screening, Presymptomatically Identified Infant With Late-Onset Pompe Disease: Case Report, Parental Experience, and Recommendations. Int J Neonatal Screen. 2020; 6(1): 22. Published 2020 Mar 14.
In contrast, late-onset Pompe disease (LOPD) patients possess some residual GAA enzymatic activity, which slows disease progression. Often discovered in adults after years of proximal myopathic symptoms, LOPD patients can eventually lose skeletal muscle strength and become reliant on artificial respiration and nonambulatory without treatment
[3]
M. L. C. Hagemans, L. P. F. Winkel, P. A: Van Doorn, et al.: Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients, Brain. 128: 671-677.
Due to myopathic damage, individuals with Pompe disease often exhibit elevated levels of blood creatine phosphokinase, transaminases, and LDH. While these tests, along with urine hexose tetrasaccharide (a marker of glycogen buildup), can indicate the presence of Pompe disease, they are neither sensitive nor specific. Diagnosis is confirmed by detecting deficient GAA enzymatic activity in white blood cells and identifying biparentally inherited GAA mutations
[4]
Bali DS, Goldstein JL, Banugaria S, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012; 160C(1): 40-49.
. According to one study, the overall prevalence of Pompe disease is approximately one in 40,000, with an incidence of one in 38,000 for infantile GSD II and one in 57,000 for adult GSD II
[5]
Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999; 7(6): 713-716.
Intravenous enzyme replacement therapy (ERT) with recombinant human GAA (rhGAA) has significantly altered the course of Pompe disease. The efficacy of rhGAA ERT has been thoroughly demonstrated for both IOPD and LOPD. In IOPD patients, overall survival and ventilation-free survival rates have significantly improved, with early initiation of rhGAA treatment being linked to better developmental and survival outcomes
[6]
Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. 2015; 166(4): 985-91. e912.
. Given that cross-reactive immunologic material (CRIM) status and immune response are crucial factors in treatment outcomes, pre-rhGAA immunomodulation is recommended to mitigate the neutralizing effect of the anti-rhGAA immune response
[7]
Desai AK, Li C, Rosenberg AS, Kishnani PS. Immunological challenges and approaches to immunomodulation in Pompe disease: a literature review. Ann Transl Med. 2019; 7(13): 285.
. For LOPD patients, rhGAA treatment has led to improved ambulatory and pulmonary function, although reductions in the 6-minute walk test and pulmonary function tests tend to appear later in the disease progression
[8]
Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017; 264(4): 621-630.
Here is a case of an 11-year-old boy with a nine-year history of progressive muscle weakness, recent anemia and heart failure, and a family history of consanguineous marriages, who presented with clinical, radiological, and laboratory findings suggestive of late-onset Pompe disease, potentially the second reported case in Pakistan.
2. Case Presentation
An 11-year-old boy was reported to the pediatric emergency department with progressive muscles weakness for the last nine years and signs and symptoms of anemia (easily fatigued, shortness of breath, pale appearance) as well as heart failure (orthopnea, shortness of breath, and dyspnea) for the last one month. According to his mother, he has trouble ascending stairs and walks with a waddling stride. He has consanguineous marriages in his family. This condition was found in the brother and maternal uncle. On examination, his blood pressure was 90/50 mmHg, his heart rate was 140 per minute regular and his respiratory rate was 38 breaths per minute. A young, pale-appearing, malnourished and with decreased muscle mass was lying on bed in a propped up position with respiratory distress and tachypnea during a general physical examination. His cardiovascular system examination reveals apex beat displacement, elevated JVP, bilateral pedal edema, mild ascites, positive hepatojagular reflux, and systolic murmurs in the mitral and tricuspid areas.
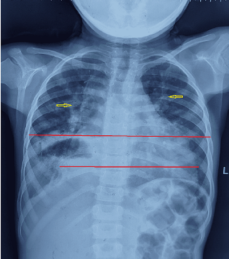
Examination of the respiratory system reveals bilateral crepitation and wheezes, as well as signs of exaggerated breathing efforts. On musculoskeletal examination, reduced muscle mass, especially proximal muscles, was more affected. Power was decreased more in proximal muscles. On abdominal examination, the liver and spleen were palpable, and there was mild ascites with shifting dullness. Other systemic reviews were unremarkable. On a chest x-ray, an enlarged cardiac shadow (red line) with pleural effusion on right side (green arrow) and few bilateral radio-opaque shadows (yellow arrow) can be seen (Figure 1). Echocardiography shows impaired left ventricle systolic function, mild to moderate mitral and tricuspid regurgitation. The diastolic dimension of the left ventricle was 5.2 cm (normal for this age, 2.8-3.5 cm), and the diastolic dimension of the right ventricle was 4.8 cm (normal for this age, 2.5-3.5 cm), Fractional shortening 20% (normal 30-44%), Ejection fraction 35% (normal >50%); the remainder of the echo results were normal. Abdominal ultrasound shows hepatic enlargement, size of 18 cm (normal 7.5-13.5 cm), splenomegaly of 15 cm (normal 7.3-11.3 cm), inferior vena cava and intrahepatic veins dilated, mild abdominopelvic ascites. Aspartate aminotransferase was 101 IU/L (normal 10-50 IU/L), LDH was 376 IU/L (80-235 IU/L), serum albumin was 2.3 g/dL (3.5-5.5 g/dL), creatine kinase-MB was 40 IU/L (25 IU/L), creatine kinase was 728 IU/L (0-189 IU/L).
Figure 1. Chest x-rays shows an enlarged cardiac shadow (red line) with pleural effusion on right side (green arrow) and few bilateral radio-opaque shadows (yellow arrow).
A complete blood count showed normal white blood cell and platelet counts with a decreased red blood cell count. Hemoglobin was 1.81 million cells per mcL (4.66 million cells per mcL).4.81 g/dL (normal 11.5-17.5 g/dL). A peripheral smear shows normocytic, normochromic anemia. Other investigations were normal. Muscle biopsy was taken from hamstring muscles, which showed PAS stain positive granules. All these clinical, radiological and laboratory findings are suggestive of Pompe’s disease.
3. Discussion
Pompe's disease is an autosomal recessive disease caused by an insufficiency in acid alpha-glucosidase, which results in intra-lysosomal glycogen buildup and muscle failure
. Hypertrophic cardiomyopathy and significant widespread weakness are the hallmarks of infantile-onset Pompe disease, which appears in the first few months of birth and progresses quickly, with mortality generally happening by the age of one year
[10]
Dasouki M, Jawdat O, Almadhoun O, Pasnoor M, McVey AL, Abuzinadah A, Herbelin L, Barohn RJ, Dimachkie MM. Pompe disease: literature review and case series. Neurol Clin. 2014 Aug; 32(3): 751-76, ix.
. Late-onset Pompe's disease is distinguished by the beginning of symptoms beyond the age of one year, the lack or mildness of heart involvement, and a slower progression of symptoms, which are mostly associated to respiratory muscle involvement and increasing skeletal muscle dysfunction
[11]
Case LE, Kishnani PS. Physical therapy management of Pompe disease. Genet Med. 2006; 8(5): 318-327.
Our patient had a late-onset condition, because he appeared at 11 years old with a 9-year history of gradual skeletal muscle weakening, signs and symptoms of anemia, and heart failure over the previous month. Recent Clinical studies of enzyme replacement treatments have started to offer more opportunities for improvement in motor status, performance, and survival than it has ever been, with the goal of improving physical health in Pompe's disease patients. Children survive longer, with few completing milestones such as independent sitting, crawling, and walking that are seldom accomplished in the untreated natural course of the condition
[12]
Pfrimmer C, Smitka M, Muschol N, Husain RA, Huemer M, Hennermann JB, Schuler R, Hahn A. Long-Term Outcome of Infantile Onset Pompe Disease Patients Treated with Enzyme Replacement Therapy - Data from a German-Austrian Cohort. J Neuromuscul Dis. 2024; 11(1): 167-177.
Chest X rays showed enlarge cardiac shadow. Echocardiography shows impaired LV systolic function with mild to moderate tricuspid and mitral regurgitation and increased cardiac dimensions. Ultrasound shows massive hepatosplenomegaly. Late-onset pompe disease is a debilitating myopathy that progresses over time. There is currently no therapy that has been shown to be effective in clinical trials. In the future, enzyme replacement therapy may be effective, but it will be costly and not readily available
[13]
Rovelli V, Zuvadelli J, Piotto M, et al. L-alanine supplementation in Pompe disease (IOPD): a potential therapeutic implementation for patients on ERT? A case report. Ital J Pediatr. 2022; 48(1): 48. Published 2022 Mar 28.
Some evidence suggests that strong-protein diet may be beneficial for adult-onset illnesses
[14]
Golsari A, Nasimzadah A, Thomalla G, Keller S, Gerloff C, Magnus T. Prevalence of adult Pompe disease in patients with proximal myopathic syndrome and undiagnosed muscle biopsy. Neuromuscul Disord. 2018; 28(3): 257-261.
. Mechanical ventilation is generally required in both children and adults with conditions that advance to respiratory failure. Noninvasive ventilation during sleep may help certain adult-onset diseases with overnight hypoxemia and daytime hypercapnia
[15]
Mellies U, Stehling F, Dohna-Schwake C, Ragette R, Teschler H, Voit T. Respiratory failure in Pompe disease: treatment with noninvasive ventilation. Neurology. 2005; 64(8): 1465-1467.
This case report highlights an 11-year-old boy with late-onset Pompe disease, presenting with progressive muscle weakness, anemia, and unexpected heart failure. These findings, along with radiological and laboratory data, support the diagnosis of Pompe disease. This case emphasizes the variability in clinical presentation and the importance of considering Pompe disease in differential diagnoses for progressive myopathies in children. Early diagnosis and intervention, including enzyme replacement therapy, are crucial for improving outcomes. As awareness of Pompe disease grows, further studies and case reports will enhance our understanding of this rare condition and its management in diverse populations.
Wang RY. A Newborn Screening, Presymptomatically Identified Infant With Late-Onset Pompe Disease: Case Report, Parental Experience, and Recommendations. Int J Neonatal Screen. 2020; 6(1): 22. Published 2020 Mar 14.
M. L. C. Hagemans, L. P. F. Winkel, P. A: Van Doorn, et al.: Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients, Brain. 128: 671-677.
Bali DS, Goldstein JL, Banugaria S, et al. Predicting cross-reactive immunological material (CRIM) status in Pompe disease using GAA mutations: lessons learned from 10 years of clinical laboratory testing experience. Am J Med Genet C Semin Med Genet. 2012; 160C(1): 40-49.
Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in The Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet. 1999; 7(6): 713-716.
Chien YH, Lee NC, Chen CA, et al. Long-term prognosis of patients with infantile-onset Pompe disease diagnosed by newborn screening and treated since birth. J Pediatr. 2015; 166(4): 985-91. e912.
Desai AK, Li C, Rosenberg AS, Kishnani PS. Immunological challenges and approaches to immunomodulation in Pompe disease: a literature review. Ann Transl Med. 2019; 7(13): 285.
Schoser B, Stewart A, Kanters S, et al. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: a systematic review and meta-analysis. J Neurol. 2017; 264(4): 621-630.
Dasouki M, Jawdat O, Almadhoun O, Pasnoor M, McVey AL, Abuzinadah A, Herbelin L, Barohn RJ, Dimachkie MM. Pompe disease: literature review and case series. Neurol Clin. 2014 Aug; 32(3): 751-76, ix.
Pfrimmer C, Smitka M, Muschol N, Husain RA, Huemer M, Hennermann JB, Schuler R, Hahn A. Long-Term Outcome of Infantile Onset Pompe Disease Patients Treated with Enzyme Replacement Therapy - Data from a German-Austrian Cohort. J Neuromuscul Dis. 2024; 11(1): 167-177.
Rovelli V, Zuvadelli J, Piotto M, et al. L-alanine supplementation in Pompe disease (IOPD): a potential therapeutic implementation for patients on ERT? A case report. Ital J Pediatr. 2022; 48(1): 48. Published 2022 Mar 28.
Golsari A, Nasimzadah A, Thomalla G, Keller S, Gerloff C, Magnus T. Prevalence of adult Pompe disease in patients with proximal myopathic syndrome and undiagnosed muscle biopsy. Neuromuscul Disord. 2018; 28(3): 257-261.
Rizwanullah, Mulakalapalli, S., Bassi, R., Patel, H., Khan, W. (2024). A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure. American Journal of Pediatrics, 10(3), 132-135. https://doi.org/10.11648/j.ajp.20241003.15
Rizwanullah; Mulakalapalli, S.; Bassi, R.; Patel, H.; Khan, W. A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure. Am. J. Pediatr.2024, 10(3), 132-135. doi: 10.11648/j.ajp.20241003.15
Rizwanullah, Mulakalapalli S, Bassi R, Patel H, Khan W. A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure. Am J Pediatr. 2024;10(3):132-135. doi: 10.11648/j.ajp.20241003.15
@article{10.11648/j.ajp.20241003.15,
author = {Rizwanullah and Srichand Mulakalapalli and Radhika Bassi and Henna Patel and Waqar Khan},
title = {A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure
},
journal = {American Journal of Pediatrics},
volume = {10},
number = {3},
pages = {132-135},
doi = {10.11648/j.ajp.20241003.15},
url = {https://doi.org/10.11648/j.ajp.20241003.15},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajp.20241003.15},
abstract = {Pompe disease is a hereditary lysosomal storage disorder characterized by a deficiency in the acid alpha-glucosidase (GAA) enzyme, leading to glycogen accumulation in muscle and neurons. Clinical manifestations vary from severe infantile-onset with hypertrophic cardiomyopathy and early mortality due to respiratory insufficiency to late-onset with proximal muscle weakness, gross motor delay, and progressive respiratory insufficiency. A case of an 11-year-old boy who reported to the pediatric emergency department with a nine-year history of progressive muscle weakness and a one-month history of anemia symptoms (easy fatigue, shortness of breath, pale appearance) and heart failure (orthopnea, dyspnea). His family history included consanguineous marriages and similar conditions in his brother and maternal uncle. On examination, he appeared pale, malnourished, and exhibited signs of respiratory distress and tachypnea. His cardiovascular examination revealed apex beat displacement, elevated JVP, bilateral pedal edema, mild ascites, positive hepatojugular reflux, and systolic murmurs. Respiratory examination indicated bilateral crepitation and wheezes. Musculoskeletal examination showed decreased muscle mass and power, especially in proximal muscles. Abdominal examination revealed hepatosplenomegaly and mild ascites. Radiological findings included an enlarged cardiac shadow with pleural effusion and bilateral radio-opaque shadows on chest x-ray, while echocardiography showed impaired left ventricular systolic function with mild to moderate mitral and tricuspid regurgitation. Laboratory tests indicated elevated aspartate aminotransferase, LDH, and creatine kinase levels, along with normocytic, normochromic anemia. Muscle biopsy from the hamstring revealed PAS stain positive granules. These clinical, radiological, and laboratory findings strongly suggest late-onset Pompe disease, marking this as potentially the second reported case in Pakistan.
},
year = {2024}
}
TY - JOUR
T1 - A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure
AU - Rizwanullah
AU - Srichand Mulakalapalli
AU - Radhika Bassi
AU - Henna Patel
AU - Waqar Khan
Y1 - 2024/08/15
PY - 2024
N1 - https://doi.org/10.11648/j.ajp.20241003.15
DO - 10.11648/j.ajp.20241003.15
T2 - American Journal of Pediatrics
JF - American Journal of Pediatrics
JO - American Journal of Pediatrics
SP - 132
EP - 135
PB - Science Publishing Group
SN - 2472-0909
UR - https://doi.org/10.11648/j.ajp.20241003.15
AB - Pompe disease is a hereditary lysosomal storage disorder characterized by a deficiency in the acid alpha-glucosidase (GAA) enzyme, leading to glycogen accumulation in muscle and neurons. Clinical manifestations vary from severe infantile-onset with hypertrophic cardiomyopathy and early mortality due to respiratory insufficiency to late-onset with proximal muscle weakness, gross motor delay, and progressive respiratory insufficiency. A case of an 11-year-old boy who reported to the pediatric emergency department with a nine-year history of progressive muscle weakness and a one-month history of anemia symptoms (easy fatigue, shortness of breath, pale appearance) and heart failure (orthopnea, dyspnea). His family history included consanguineous marriages and similar conditions in his brother and maternal uncle. On examination, he appeared pale, malnourished, and exhibited signs of respiratory distress and tachypnea. His cardiovascular examination revealed apex beat displacement, elevated JVP, bilateral pedal edema, mild ascites, positive hepatojugular reflux, and systolic murmurs. Respiratory examination indicated bilateral crepitation and wheezes. Musculoskeletal examination showed decreased muscle mass and power, especially in proximal muscles. Abdominal examination revealed hepatosplenomegaly and mild ascites. Radiological findings included an enlarged cardiac shadow with pleural effusion and bilateral radio-opaque shadows on chest x-ray, while echocardiography showed impaired left ventricular systolic function with mild to moderate mitral and tricuspid regurgitation. Laboratory tests indicated elevated aspartate aminotransferase, LDH, and creatine kinase levels, along with normocytic, normochromic anemia. Muscle biopsy from the hamstring revealed PAS stain positive granules. These clinical, radiological, and laboratory findings strongly suggest late-onset Pompe disease, marking this as potentially the second reported case in Pakistan.
VL - 10
IS - 3
ER -
Rizwanullah, Mulakalapalli, S., Bassi, R., Patel, H., Khan, W. (2024). A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure. American Journal of Pediatrics, 10(3), 132-135. https://doi.org/10.11648/j.ajp.20241003.15
Rizwanullah; Mulakalapalli, S.; Bassi, R.; Patel, H.; Khan, W. A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure. Am. J. Pediatr.2024, 10(3), 132-135. doi: 10.11648/j.ajp.20241003.15
Rizwanullah, Mulakalapalli S, Bassi R, Patel H, Khan W. A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure. Am J Pediatr. 2024;10(3):132-135. doi: 10.11648/j.ajp.20241003.15
@article{10.11648/j.ajp.20241003.15,
author = {Rizwanullah and Srichand Mulakalapalli and Radhika Bassi and Henna Patel and Waqar Khan},
title = {A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure
},
journal = {American Journal of Pediatrics},
volume = {10},
number = {3},
pages = {132-135},
doi = {10.11648/j.ajp.20241003.15},
url = {https://doi.org/10.11648/j.ajp.20241003.15},
eprint = {https://article.sciencepublishinggroup.com/pdf/10.11648.j.ajp.20241003.15},
abstract = {Pompe disease is a hereditary lysosomal storage disorder characterized by a deficiency in the acid alpha-glucosidase (GAA) enzyme, leading to glycogen accumulation in muscle and neurons. Clinical manifestations vary from severe infantile-onset with hypertrophic cardiomyopathy and early mortality due to respiratory insufficiency to late-onset with proximal muscle weakness, gross motor delay, and progressive respiratory insufficiency. A case of an 11-year-old boy who reported to the pediatric emergency department with a nine-year history of progressive muscle weakness and a one-month history of anemia symptoms (easy fatigue, shortness of breath, pale appearance) and heart failure (orthopnea, dyspnea). His family history included consanguineous marriages and similar conditions in his brother and maternal uncle. On examination, he appeared pale, malnourished, and exhibited signs of respiratory distress and tachypnea. His cardiovascular examination revealed apex beat displacement, elevated JVP, bilateral pedal edema, mild ascites, positive hepatojugular reflux, and systolic murmurs. Respiratory examination indicated bilateral crepitation and wheezes. Musculoskeletal examination showed decreased muscle mass and power, especially in proximal muscles. Abdominal examination revealed hepatosplenomegaly and mild ascites. Radiological findings included an enlarged cardiac shadow with pleural effusion and bilateral radio-opaque shadows on chest x-ray, while echocardiography showed impaired left ventricular systolic function with mild to moderate mitral and tricuspid regurgitation. Laboratory tests indicated elevated aspartate aminotransferase, LDH, and creatine kinase levels, along with normocytic, normochromic anemia. Muscle biopsy from the hamstring revealed PAS stain positive granules. These clinical, radiological, and laboratory findings strongly suggest late-onset Pompe disease, marking this as potentially the second reported case in Pakistan.
},
year = {2024}
}
TY - JOUR
T1 - A Rare Case of Late Onset-Pompe’s Disease: Presented as Heart Failure
AU - Rizwanullah
AU - Srichand Mulakalapalli
AU - Radhika Bassi
AU - Henna Patel
AU - Waqar Khan
Y1 - 2024/08/15
PY - 2024
N1 - https://doi.org/10.11648/j.ajp.20241003.15
DO - 10.11648/j.ajp.20241003.15
T2 - American Journal of Pediatrics
JF - American Journal of Pediatrics
JO - American Journal of Pediatrics
SP - 132
EP - 135
PB - Science Publishing Group
SN - 2472-0909
UR - https://doi.org/10.11648/j.ajp.20241003.15
AB - Pompe disease is a hereditary lysosomal storage disorder characterized by a deficiency in the acid alpha-glucosidase (GAA) enzyme, leading to glycogen accumulation in muscle and neurons. Clinical manifestations vary from severe infantile-onset with hypertrophic cardiomyopathy and early mortality due to respiratory insufficiency to late-onset with proximal muscle weakness, gross motor delay, and progressive respiratory insufficiency. A case of an 11-year-old boy who reported to the pediatric emergency department with a nine-year history of progressive muscle weakness and a one-month history of anemia symptoms (easy fatigue, shortness of breath, pale appearance) and heart failure (orthopnea, dyspnea). His family history included consanguineous marriages and similar conditions in his brother and maternal uncle. On examination, he appeared pale, malnourished, and exhibited signs of respiratory distress and tachypnea. His cardiovascular examination revealed apex beat displacement, elevated JVP, bilateral pedal edema, mild ascites, positive hepatojugular reflux, and systolic murmurs. Respiratory examination indicated bilateral crepitation and wheezes. Musculoskeletal examination showed decreased muscle mass and power, especially in proximal muscles. Abdominal examination revealed hepatosplenomegaly and mild ascites. Radiological findings included an enlarged cardiac shadow with pleural effusion and bilateral radio-opaque shadows on chest x-ray, while echocardiography showed impaired left ventricular systolic function with mild to moderate mitral and tricuspid regurgitation. Laboratory tests indicated elevated aspartate aminotransferase, LDH, and creatine kinase levels, along with normocytic, normochromic anemia. Muscle biopsy from the hamstring revealed PAS stain positive granules. These clinical, radiological, and laboratory findings strongly suggest late-onset Pompe disease, marking this as potentially the second reported case in Pakistan.
VL - 10
IS - 3
ER -