1. Introduction
The extensive use of fossil fuels has exacerbated the greenhouse effect and severe climate warming. Compared to coal and oil, natural gas possesses high energy density and lower carbon emissions, making it a crucial transitional energy source in the shift toward a low-carbon energy structure
. However, how to store and transport natural gas more efficiently remains an urgent issue requiring exploration
.
Natural gas hydrates are non-stoichiometric cage-like crystal complexes formed by intermolecular interactions between water molecules
. They possess an exceptionally high gas storage capacity per unit volume (164 v/v STP)
. Furthermore, vast reserves of natural gas hydrates have been identified in marine continental slopes and terrestrial permafrost layers
, demonstrating the practical feasibility of hydrate formation for natural gas storage and transportation
.
The hydrate method requires only moderate temperature and pressure conditions (274.3K, 10MPa) for natural gas storage and transportation
. This contrasts sharply with liquefied natural gas (LNG) technology, which demands extremely low temperatures, and compressed natural gas (CNG), which necessitates high pressures
. Achieving these extreme conditions consumes substantial energy resources. Therefore, the hydrate method offers greater energy efficiency and lower carbon emissions while providing comparable gas storage capacity, making it a highly promising approach
. However, current hydrate formation is constrained by the challenges of a long nucleation induction time, slow growth rate, and the thermodynamically demanding temperature-pressure environment, preventing its industrial application
| [10] | Y. He, M.-T. Sun, C. Chen, G.-D. Zhang, K. Chao, Y. Lin and F. Wang, Journal of Materials Chemistry A 2019, 7, 21634-21661. https://doi.org/10.1039/c9ta07071k |
| [11] | Z. Xia, Q. Zhao, Z. Chen, X. Li, Y. Zhang, C. Xu and K. Yan, Journal of Natural Gas Science and Engineering 2022, 101, 104528. https://doi.org/10.1016/j.jngse.2022.104528 |
| [12] | Y. Feng, Y. Han, P. Gao, Y. Kuang, L. Yang, J. Zhao and Y. Song, Energy 2024, 290, 130228.
https://doi.org/10.1016/j.energy.2023.130228 |
| [13] | L. Yang, K. J. Shi, A. X. Qu, H. Y. Liang, Q. P. Li, X. Lv, S. D. Leng, Y. Z. Liu, L. X. Zhang, Y. Liu, B. Xiao, S. X. Yang, J. F. Zhao and Y. C. Song, Energy 2023, 276.
https://doi.org/10.1016/j.energy.2023.127545 |
[10-13]
.
Researchers have conducted extensive experiments employing various thermodynamic and kinetic promotion methods to enhance hydrate nucleation and growth
| [14] | X. N. Ma, Y. Y. Liu, Y. Cheng, S. W. Zhang, J. Yan, L. L. Jiang, Y. Zhang and Y. C. Song, Journal of Environmental Chemical Engineering 2025, 13.
https://doi.org/10.1016/j.jece.2025.120155 |
| [15] | Y. Zhao, M. Z. Yang, M. Li, H. S. Dong, Y. Ge, Q. P. Li, L. X. Zhang, Y. Liu, L. Yang, Y. C. Song and J. F. Zhao, Acs Applied Materials & Interfaces 2022, 14, 33141-33150.
https://doi.org/10.1021/acsami.2c06230 |
| [16] | H. Q. Liu, S. Wang, C. R. Shi, Y. C. Song, Y. X. Fu, Z. Li, L. X. Zhang, C. Chen, M. J. Yang and Z. Ling, Fuel 2025, 381.
https://doi.org/10.1016/j.fuel.2024.133291 |
| [17] | H. Q. Liu, C. R. Shi, S. Wang, L. X. Zhang, J. F. Zhao, M. J. Yang, C. Chen, Y. C. Song and Z. Ling, Journal of Colloid and Interface Science 2023, 641, 812-819.
https://doi.org/10.1016/j.jcis.2023.03.118 |
| [18] | Z. C. Cheng, L. T. Sun, Y. Y. Liu, L. L. Jiang, B. B. Chen and Y. C. Song, Renewable Energy 2023, 218.
https://doi.org/10.1016/j.renene.2023.119350 |
| [19] | H. Z. Xu, Y. Y. Liu, S. Y. He, J. N. Zheng, L. L. Jiang and Y. C. Song, Energy 2024, 291.
https://doi.org/10.1016/j.energy.2024.130280 |
| [20] | Y. Y. Liu, L. T. Sun, J. N. Ren, T. Yu, L. L. Jiang and Y. C. Song, Journal of Energy Chemistry 2025, 108, 635-644.
https://doi.org/10.1016/j.jechem.2025.04.064 |
| [21] | L. J. Sun, H. L. Sun, C. Y. Yuan, L. X. Zhang, L. Yang, Z. Ling, J. F. Zhao and Y. C. Song, Chemical Engineering Journal 2023, 454. https://doi.org/10.1016/j.cej.2022.140325 |
| [22] | Y. Wei, J. Worley, L. E. Zerpa, Y. C. Chien, D. Dunn-Rankin, M. T. Kezirian and C. A. Koh, Fluid Phase Equilibria 2025, 591. https://doi.org/10.1016/j.fluid.2024.114286 |
[14-22]
. After nucleation, water molecules combine with gas molecules to form progressively larger clusters until reaching a critical size. Once entering the sustained growth phase, the process is governed by two factors: mass transfer of hydrate-forming components to the growing crystal and heat transfer from the heat released during crystal formation
| [23] | C. A. Koh, E. D. Sloan, A. K. Sum and D. T. Wu, Fundamentals and Applications of Gas Hydrates, 2011, 2, 237-257.
https://doi.org/10.1146/annurev-chembioeng-061010-114152 |
| [24] | Z. Yin, M. Khurana, H. K. Tan and P. Linga, Chemical Engineering Journal 2018, 342, 9-29.
https://doi.org/10.1016/j.cej.2018.01.120 |
[23, 24]
. In recent years, tetrahydrofuran (THF)
| [25] | H. P. Veluswamy, A. J. H. Wong, P. Babu, R. Kumar, S. Kulprathipanja, P. Rangsunvigit and P. Linga, Chemical Engineering Journal 2016, 290, 161-173.
https://doi.org/10.1016/j.cej.2016.01.026 |
| [26] | D. Mech, P. Gupta and J. S. Sangwai, Journal of Natural Gas Science and Engineering 2016, 35, 1519-1534.
https://doi.org/10.1016/j.jngse.2016.06.013 |
[25, 26]
, cyclopentane (CP)
, and tetrabutylammonium bromide (TBAB)
| [29] | V. R. Avula, R. L. Gardas and J. S. Sangwai, Journal of Natural Gas Science and Engineering 2016, 33, 509-517.
https://doi.org/10.1016/j.jngse.2016.05.051 |
| [30] | Z. C. Cheng, L. T. Sun, Y. Y. Liu, H. Z. Xu, L. L. Jiang, L. Wang and Y. C. Song, Energy 2023, 280.
https://doi.org/10.1016/j.energy.2023.128141 |
[29, 30]
have been widely employed as thermodynamic promoters to mitigate the temperature and pressure requirements for hydrate growth. However, conventional thermodynamic promoters exhibit toxicity and carcinogenicity, posing environmental and biological risks that necessitate limiting their extensive use
. Therefore, we can simultaneously introduce kinetic promotion methods (mechanical, chemical), such as adding kinetic promoters, to enhance the promotion effect and thereby reduce the required amount of thermodynamic promoters
| [32] | N. Xu, Y. Liu, Z. C. Cheng, S. J. Wang, L. L. Jiang and Y. C. Song, Energy & Fuels 2020, 34, 7307-7315.
https://doi.org/10.1021/acs.energyfuels.0c00773 |
| [33] | Q. Zhang, J. J. Zheng, B. Y. Zhang and P. Linga, Energy 2023, 274. https://doi.org/10.1016/j.energy.2023.127322 |
[32, 33]
.
Selecting appropriate kinetic promoters is crucial. These include chemical surfactants like SDS, which significantly improve methane hydrate formation kinetics
| [32] | N. Xu, Y. Liu, Z. C. Cheng, S. J. Wang, L. L. Jiang and Y. C. Song, Energy & Fuels 2020, 34, 7307-7315.
https://doi.org/10.1021/acs.energyfuels.0c00773 |
| [34] | N. S. Molokitina, A. N. Nesterov, L. S. Podenko and A. M. Reshetnikov, Fuel 2019, 235, 1400-1411.
https://doi.org/10.1016/j.fuel.2018.08.126 |
[32, 34]
. Proposed promotion mechanisms encompass capillary theory, micelle theory, and adsorption theory, but no consensus has been reached yet
| [35] | Y. Zhong and R. E. Rogers, Chemical Engineering Science 2000, 55, 4175-4187.
https://doi.org/10.1016/S0009-2509(00)00072-5 |
| [36] | W. Lin, G. J. Chen, C. Y. Sun, X. Q. Guo, Z. K. Wu, M. Y. Liang, L. T. Chen and L. Y. Yang, Chemical Engineering Science 2004, 59, 4449-4455. https://doi.org/10.1016/j.ces.2004.07.010 |
| [37] | J. S. Zhang, C. Lo, P. Somasundaran and J. W. Lee, Journal of Colloid and Interface Science 2010, 341, 286-288.
https://doi.org/10.1016/j.jcis.2009.09.052 |
[35-37]
. However, chemical agents like SDS may harm aquatic organisms upon entering water bodies, posing potential risks to ecosystems
. Consequently, green, eco-friendly, and biocompatible biosurfactants—such as rhamnolipids—have gained research attention
| [39] | S. Jadav, N. Sakthipriya, M. Doble and J. S. Sangwai, Journal of Natural Gas Science and Engineering 2017, 43, 156-166.
https://doi.org/10.1016/j.jngse.2017.03.032 |
| [40] | Y. Zhang, J. R. Zhang, X. G. Xu, W. T. Liu, Y. S. Xu, M. J. Yang and Y. C. Song, Fuel 2024, 357.
https://doi.org/10.1016/j.fuel.2023.129873 |
[39, 40]
. Yet both chemical and biosurfactants face criticism due to the extensive bubble formation during hydrate decomposition, complicating cleanup. Additionally, nanoparticles including carbon nanotubes
, metal-organic frameworks (MOFs)
, metal nanoparticles
, and graphene
effectively accelerate hydrate formation kinetics. This is achieved by introducing numerous nucleation sites, increasing local methane gas concentrations, and potentially enhancing thermal conductivity to improve heat exchange and prevent rapid temperature spikes
. Although nanoparticles do not generate bubbles, their preparation processes are typically complex and costly, and their widespread use may pose potential environmental risks. Future research should focus on developing novel nanoparticles with simplified preparation processes and enhanced environmental sustainability.
In recent years, amino acids have garnered significant attention from researchers due to their environmental friendliness, high biocompatibility, bubble-free hydrate decomposition properties, and excellent hydrate kinetic promotion effects
| [46] | B. Li, Y. Y. Lu and Y. L. Li, Journal of Marine Science and Engineering 2022, 10. https://doi.org/10.3390/jmse10081134 |
| [47] | G. Bhattacharjee and P. Linga, Energy & Fuels 2021, 35, 7553-7571. https://doi.org/10.1021/acs.energyfuels.1c00502 |
| [48] | C. B. Bavoh, B. Lal, H. Osei, K. M. Sabil and H. Mukhtar, Journal of Natural Gas Science and Engineering 2019, 64, 52-71. https://doi.org/10.1016/j.jngse.2019.01.020 |
[46-48]
. Particularly hydrophobic amino acids demonstrate superior promotion effects compared to hydrophilic ones. This is because hydrophobic amino acids disperse at the gas-liquid interface, reducing surface tension and enriching gas molecules with their hydrophobic side chains, while hydrophilic amino acids tend to disperse in the aqueous phase
. Additionally, researchers have investigated the effects of peptides with varying molecular weights and macromolecular proteins on hydrate formation kinetics
| [50] | Y. X. Jia, Y. Zhao, M. Li, L. X. Zhang, Y. Z. Liu, H. S. Dong, J. F. Zhao, L. Yang and Y. C. Song, Acs Sustainable Chemistry & Engineering 2022, 10, 11320-11329.
https://doi.org/10.1021/acssuschemeng.2c03342 |
[50]
. However, most previous studies indicate that proteins or peptides inhibit hydrate growth, such as antifreeze proteins
| [51] | V. K. Walker, H. Zeng, H. Ohno, N. Daraboina, H. Sharifi, S. A. Bagherzadeh, S. Alavi and P. Englezos, Canadian Journal of Chemistry 2015, 93, 839-849.
https://doi.org/10.1139/cjc-2014-0538 |
| [52] | C. Chen, Y. Zhang, J. Y. Sun, Y. Liu, Y. Qin, Z. Ling, W. G. Liu and W. Z. Li, Fuel 2022, 327.
https://doi.org/10.1016/j.fuel.2022.125060 |
[51, 52]
and antifreeze peptides
| [53] | N. Zhang, Y. Zhu, Y. N. Li, L. R. Zhang, F. S. Zhang and J. J. Liu, Journal of Chemical Physics 2024, 161.
https://doi.org/10.1063/5.0211732 |
| [54] | S. X. Li, R. J. Lv, Z. S. Yan, F. Huang, X. R. Zhang, G. J. Chen and T. T. Yue, Acs Sustainable Chemistry & Engineering 2020, 8, 4256-4266.
https://doi.org/10.1021/acssuschemeng.9b07701 |
[53, 54]
. In contrast, Gao's research revealed that ovalbumin inhibits carbon dioxide hydrate growth at high concentrations but promotes it at low concentrations
. Bastien Radola's work demonstrated that the TAFDGGS peptide effectively promotes methane hydrate formation, with its mechanism elucidated through high-pressure experiments and molecular dynamics simulations
. Therefore, numerous proteins and peptides in nature may potentially promote hydrate nucleation and growth. Being biologically and environmentally friendly, widely available, and requiring no preparation, they could emerge as promising hydrate kinetics promoters in the near future. However, relevant research remains scarce, necessitating further investigation into their methane hydrate promotion effects under diverse environmental and experimental conditions.
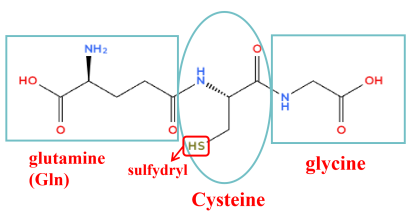
Glutathione (GSH), a tripeptide formed by the condensation of glutamic acid, cysteine, and glycine via peptide bonds (as shown in
Figure 1), is naturally abundant and readily available
| [57] | M. Knapen, P. L. M. Zusterzeel, W. H. M. Peters and E. A. P. Steegers, European Journal of Obstetrics & Gynecology and Reproductive Biology 1999, 82, 171-184.
https://doi.org/10.1016/S0301-2115(98)00242-5 |
| [58] | X. M. Guan, Medicinal Chemistry Research 2023, 32, 1972-1994. https://doi.org/10.1007/s00044-023-03116-9 |
| [59] | H. Jefferies, J. Coster, A. Khalil, J. Bot, R. D. McCauley and J. C. Hall, Anz Journal of Surgery 2003, 73, 517-522.
https://doi.org/10.1046/j.1445-1433.2003.02682.x |
[57-59]
. It is widely distributed in both animal and plant sources, including baker's yeast and wheat germ. Due to its antioxidant properties, GSH has found extensive applications in cosmetics and food industries. GSH possesses multiple strongly hydrophilic groups and relatively weak hydrophobic groups, exhibiting partial amphiphilicity. However, its strong hydrophilicity causes GSH to preferentially distribute in the aqueous phase rather than at the interfacial boundary. GSH is indeed a crucial and potent small-molecule polypeptide.
Figure 1. Schematic diagram of glutathione molecular structure, comprising glutamine, cysteine, and glycine. Possesses numerous hydrophilic groups.
This paper investigates the effects of GSH and amino acids on promoting the formation kinetics of methane hydrates. Research indicates that different concentrations of GSH, as well as composite systems of GSH and specific amino acids, promote methane hydrate formation kinetics to varying degrees. This process generates no bubbles, is safe and non-toxic, and is environmentally friendly. The primary objective is to investigate the synergistic promotion of hydrate growth by the GSH-TBAB composite system, further shortening induction time and increasing gas storage capacity. A smaller TBAB dosage achieves a hydrate promotion effect comparable to that of GSH alone. Furthermore, we proposed potential mechanisms for the synergistic promotion by GSH, amino acids, and TBAB.
2. Experimental Section
2.1. Materials and Sample Preparation
Methane gas (99.9% purity) used in the experiments was purchased from Dalian Special Gas Co., Ltd. (China). Glutathione (98% purity), leucine (99% purity), methionine (99% purity), glycine (99.5% purity), and tetrabutylammonium bromide (TBAB, 99% purity) (TBAB). Sodium chloride (NaCl, AR analytical grade) was purchased from Sinopharm Chemical Reagent Co., Ltd. (China). Deionized water (DIW) with a resistivity of 18.2 MΩcm−1 was prepared in our laboratory using an ultrapure water system (Aquapro2S, Aquapro International Company LLC, USA).
2.2. Methane Hydrate Formation Experiment
2.2.1. Experimental Equipment and Procedure
Schematic diagram of the methane hydrate formation experimental system (
Figure 1). Prior to each experiment, the stainless steel high-pressure reactor was thoroughly rinsed with tap water followed by deionized water at least three times. The high-pressure reactor has a volume of approximately 233 ml and is designed to withstand pressures up to 10 MPa. Temperature and pressure during the experiment were monitored using a temperature sensor (PT100) and a pressure sensor, with accuracies of ±0.01°C and ±0.01 MPa, respectively. Temperature and pressure data were recorded every 30 seconds via a data logger and stored on a computer hard drive. First, accurately weigh the required masses of GSH, amino acids, TBAB, and sodium chloride using an analytical balance (accuracy 0.0001 g). Transfer these to a 150 ml beaker, add deionized water, and process with ultrasonication (90 W, 40 kHz, approximately 23°C) to obtain 120 g of uniformly mixed solution. After the reactor dried, the solution was transferred into the reactor, tightly sealed, and subjected to magnetic stirring at 300 rpm to accelerate the reaction. Rotor agitation increased the solution's Reynolds number, enhancing fluid flow and heat transfer. This expanded the contact area between methane gas and water, shortening the induction time in pure water conditions and reducing experimental randomness
. The reactor was then placed in a water-glycol bath (Ployscience, USA) for temperature control. Methane gas was repeatedly purged through the reactor at least three times to remove residual air. For experiments without the thermodynamic promoter TBAB, the initial pressure was set to 8.0 MPa. For experiments with TBAB, the pressure was set to 6.0 MPa. Specifically, when the temperature stabilized at 15°C, methane gas was introduced at 6 MPa or 8 MPa, respectively, The experimental parameters for different conditions as shown in
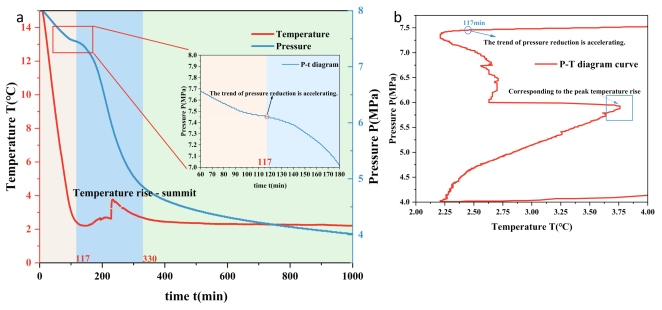
Table 1. The system was then held for at least one hour to ensure temperature and pressure values stabilized at the set points, eliminating the influence of solubility on the experiment. The water bath program was set to decrease the bath temperature at a constant rate to approximately 1°C below the preset temperature. When the reactor temperature dropped below the corresponding phase equilibrium conditions, hydrate nucleation might occur within the reactor. This was confirmed by sudden temperature increases or pressure drops. The temperature-pressure curve for hydrate formation over time is shown in
Figure 2. Each experimental run lasted 1000 minutes to ensure complete hydrate formation. To avoid the influence of random hydrate formation on the experimental results, each set of experiments was repeated.
Table 1. The experimental parameters for different conditions.
sample | P/MPa | T/K |
Pure water | 8.0 | 274.15 |
GSH 300ppm |
GSH 500ppm |
GSH 1000ppm |
GSH 2000ppm |
GSH 2000ppm + methionine |
GSH 2000ppm + glycine |
GSH 2000ppm + leucine |
GSH 2000ppm + NaCl 0.5wt% |
GSH 2000ppm + NaCl 1wt% |
GSH 2000ppm + NaCl 3.5wt% |
TBAB 3wt% | 6.0 | 274.15 |
TBAB 5wt% |
TBAB 3wt% + GSH 2000ppm |
TBAB 5wt% + GSH 2000ppm |
TBAB 5wt% + GSH 2000ppm + NaCl 1wt% |
TBAB 5wt% + GSH 2000ppm + NaCl 3.5wt% |
Figure 2. (a) P-T variation during hydrate formation (using GSH 2000 ppm as an example). At 117 min, the pressure decline accelerates, indicating hydrate nucleation. Subsequently, hydrate formation intensifies rapidly within a short timeframe, causing a sharp pressure drop and significant temperature rise. (b) Clearly shows that rapid formation commences shortly after hydrate nucleation begins.
2.2.2. Calculation Method
The induction time tind defined in this study is the time interval from the initiation of the constant-rate temperature decrease from the water bath temperature (15°C) until the onset of hydrate nucleation (manifested by a noticeable pressure drop and temperature rise). The unit of induction time is min. Each data set requires replication to minimize randomness.
The amount of methane consumed (methane absorption) within a given time t is calculated using the gas law formula.
(1)
Here, the initial methane gas volume is defined as n0, the residual gas volume in the reactor at time t is defined as nt, V is the gas phase volume in the reactor, R is the universal gas constant, T is the temperature, P is the pressure in the reactor. z is the compression factor:
(2)
where Tc is the critical temperature, with a value of 190.6 K for methane; Pc is the critical pressure, with a value of 4.599 MPa for methane; is the eccentricity factor, with a value of 0.012.
(3)
is the hydration number, typically specified as 5.75; M is the molar mass of water, valued at 18 g/mol; α is the volume ratio of hydrate to converted water, set at 1.25 in this study.
Calculate the standard methane gas absorption based on the water volume at the end of each experiment.
(4)
where Δnend is the methane gas consumption at the end of each experimental run, and nwater is the amount of deionized water in the reactor.
Define the standard volume of methane gas stored per unit volume as the methane absorption capacity (S).
(5)
The molar volume of gas under standard conditions is 22400 cm³/mol. is the volume of hydrate at time t; is the volume of converted water at time t. This study adopted constant-volume test conditions and had a relatively high moisture content. It can be anticipated that the S value will be lower compared to the constant-pressure rich gas test conditions. Therefore, comparisons were mainly conducted under the same test conditions to highlight the remarkable promoting effect.
3. Results and Discussion
3.1. Effects of Different Concentrations of GSH on Hydrate Formation
GSH is not a surfactant but a tripeptide with weak hydrophobicity and strong hydrophilicity. Its strongly hydrophilic and weakly hydrophobic nature renders it unsuitable for critical micelle theory. Possessing only weak amphiphilicity, it does not preferentially stabilize at the gas-liquid interface like SDS but instead dissolves preferentially in aqueous solutions. Consequently, GSH's mechanism of action differs significantly from SDS. It is therefore necessary to investigate GSH's influence on methane hydrate nucleation and growth, along with its underlying mechanism. Hydrate formation kinetics can be reflected by indicators such as nucleation induction time and methane uptake.
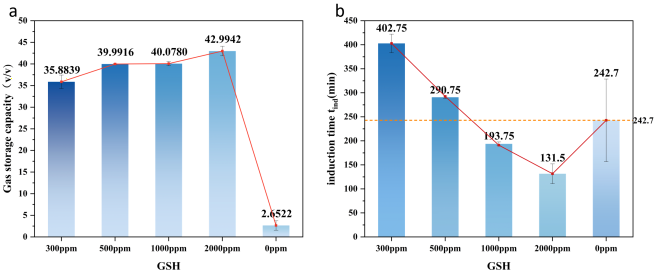
Figure 3(a) shows that the induction time first increases sharply and then gradually decreases with increasing GSH concentration. At 300 ppm, the induction time surged abruptly, extending by 160.05 min (65.9%), indicating severe inhibition of hydrate nucleation at this concentration. However, as concentration further increased, the induction time decreased. At 1000 ppm, it fell below that of pure water, indicating enhanced hydrate nucleation. At 2000 ppm, hydrate nucleation kinetics were further accelerated, with the induction time reduced by 45.8% compared to pure water and by 67.3% compared to 300 ppm.
As shown in
Figure 3(b), the methane absorption capacity (S) exhibits distinct trends. With increasing concentration, S gradually rises, showing the largest increase between 0 and 500 ppm. Beyond 500 ppm, the effect of concentration growth diminishes. At 2000 ppm, S reaches its maximum value, increasing by 1517.9% compared to pure water, demonstrating a highly significant effect. Why do the induction time data exhibit this trend of initially sharp prolongation followed by gradual reduction, while the methane absorption capacity shows a different pattern? We will explore the underlying mechanisms.
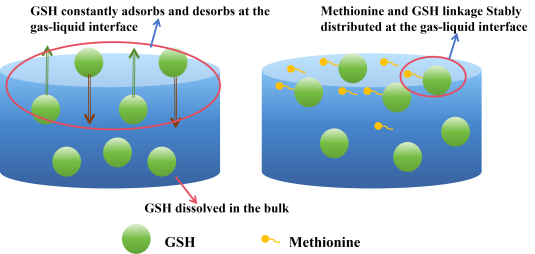
At GSH 0 ppm, due to methane's low solubility and limited gas-liquid contact area, few nucleation sites exist in the solution, making it difficult to rapidly increase local gas concentration. Consequently, even with magnetic stirring, the induction time remains prolonged under pure water conditions. With the addition of GSH particles, GSH exhibits strong hydrophilicity due to multiple hydrophilic groups (carboxyl, amino, etc.), while its short hydrophobic hydrocarbon chains confer weak hydrophobicity, resulting in partial amphiphilicity. Its strong hydrophilicity prevents GSH from forming a stable adsorption layer at the gas-liquid interface, making it unable to distribute stably at the interface. Consequently, GSH may dynamic distribute at the gas-liquid interface. However, its strong hydrophilicity causes the hydrophilic groups to be attracted by water, immersing downward into the aqueous phase. While the hydrophobic portion is repelled by water, its weak hydrophobicity allows only a small fraction to extend toward the gas phase. The nature of GSH molecules prevents them from stably distributing at the gas-liquid interface to form a dense SDS-like film, unlike SDS. Thus, GSH molecules dynamically and randomly adsorb and desorb at the gas-liquid interface due to hydrophilic and hydrophobic interactions.
Figure 3. (a) shows that the induction time of GSH decreases linearly with increasing concentration. At 300 ppm and 500 ppm, the induction time is longer than in pure water, indicating nucleation inhibition. Subsequently, it shifts to nucleation promotion, with the shortest induction time observed at 2000 ppm. (b) Gas storage capacity S increases linearly with concentration, also peaking at 2000 ppm. S is significantly higher than in pure water at all concentrations.
When the GSH concentration reaches 300 ppm, the induction time exhibits a steep increase, reaching the maximum value shown in the
Figure 3(a), as GSH dynamically adsorbs at the interface. At this point, the interface is covered by a large number of GSH molecules, forming a GSH film. However, due to its strong hydrophilicity, GSH molecules continuously adsorb and desorb from the interface. This prevents the GSH film from becoming dense and does not significantly increase the resistance to gas diffusion into the aqueous phase. Nevertheless, the strongly hydrophilic groups of GSH compete with methane gas for water molecules near the interface to form a hydration shell. This reduces the number of free water molecules, increases viscosity near the interface, and enhances the diffusion resistance of gas into water, preventing water molecules from forming ordered cage-like structures. These factors significantly raise the nucleation energy barrier for methane hydrate, prolonging the nucleation induction time.
Despite this pronounced nucleation inhibition, the methane gas uptake (S) increases rapidly, indicating that the underlying mechanisms for hydrate nucleation and growth differ. With ample growth time (1000 min), GSH dynamically adsorbs to partially reduce interfacial tension. The non-dense GSH film improves diffusion resistance for gas molecules moving from the aqueous phase to the hydrate surface. After prolonged nucleation, hydrates begin growing more rapidly under GSH influence, ultimately increasing the S value.
Since the induction time is longest at 300 ppm, further increasing GSH concentration reduces the induction time. Therefore, a nucleation transition point exists between 300 and 500 ppm. At this point, the number of GSH molecules at the interface reaches saturation, maximizing interfacial inhibition. Additional GSH can only enter the aqueous phase, providing abundant nucleation sites and adsorbing large amounts of methane molecules. This rapidly increases local gas concentration, promoting nucleation. Consequently, beyond the inflection point, interfacial inhibition no longer increases, while the promoting effect of the aqueous phase significantly enhances, causing the induction time to decrease rapidly and reach its minimum at 2000 ppm.
Simultaneously, increasing GSH in the bulk phase partially reduces surface tension and diffusion resistance while providing abundant methane adsorption sites. This enables more complete and rapid hydrate growth. Beyond 500 ppm, the upward trend in S values slows, reaching an inflection point at 500 ppm where hydrate growth is maximized and gas storage capacity increases. Further concentration increases have negligible effects on enhancing gas adsorption sites. Gas storage capacity shows no significant impact, rising slowly to a maximum at 2000 ppm. This theory elucidates the underlying mechanism behind the independent variations in nucleation induction time and methane gas absorption during hydrate growth.
Integrating the performance of GSH in terms of induction time tind and gas storage capacity S, we conclude that 2000 ppm GSH exhibits the most outstanding promotion effect on hydrate formation kinetics. Consequently, subsequent experiments will utilize this 2000 ppm concentration for investigation.
3.2. Synergistic Effects of Different Amino Acids and GSH on Hydrate Formation
To further enhance hydrate nucleation and growth, we introduced synergistic effects by combining GSH with other hydrate kinetic promoters. We selected amino acids that are environmentally friendly, biocompatible, non-foaming, and exhibit excellent methane hydrate kinetic promotion effects. Amino acids with varying degrees of hydrophobicity (leucine, methionine, glycine) were incorporated into subsequent studies.
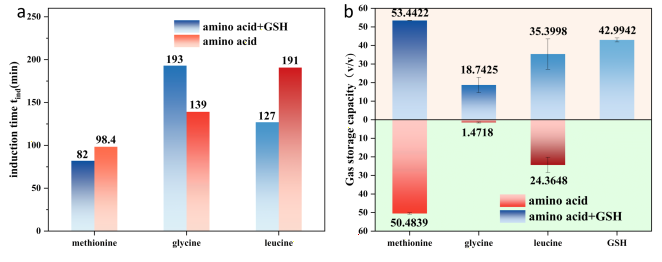
Figure 4. (a) The induction time for the methionine or leucine composite system is shorter than that for single amino acids or GSH system alone. However, the glycine complex system exhibits a significantly prolonged induction time. This indicates that hydrophobic amino acids exhibit synergistic promotion effects, while hydrophilic amino acids demonstrate synergistic inhibition. (b) The gas storage capacity (S) of all complex systems is significantly enhanced, with methionine showing the most pronounced enhancement effect, exhibiting a clear synergistic enhancement effect.
Under corresponding experimental conditions (8.0 MPa, 1°C), as shown in
Figure 4(a), the induction time for composite systems of methionine and leucine (1000 ppm) with GSH (2000 ppm) exhibited a decreasing trend compared to experiments using either amino acid or GSH alone, demonstrating a significant synergistic nucleation-promoting effect. The methionine composite system exhibited the shortest induction time at 82 min, representing a 37.6% reduction compared to the GSH-only experiment (131.5 min). However, the glycine composite system exhibited a significantly prolonged induction time (193 min), exceeding both the glycine and GSH single-component tests, indicating a synergistic inhibition phenomenon. The addition of glycine further suppressed GSH nucleation, with its induction time even approaching that of 1000 ppm GSH alone. It is intriguing that two entirely different phenomena occurred simultaneously.
By this point, as previously discussed, GSH at the gas-liquid interface has already reached saturation. A large number of GSH molecules are dispersed in the aqueous phase, forming a dynamic, unstable GSH film at the interface. The interfacial inhibition effect has peaked and will not increase further. The surface tension in the bulk phase partially decreases, accelerating gas diffusion. With numerous nucleation sites present, local gas concentrations can rise rapidly, promoting faster and more efficient hydrate nucleation and growth. Therefore, under these conditions, the primary resistance limiting the nucleation induction time is interfacial suppression. The gas-liquid interface is precisely where hydrophobic amino acids exert their predominant effect.
Due to their amphiphilic structure, hydrophobic amino acid molecules preferentially disperse at the gas-water interface. Among these, hydrophobic amino acids (leucine, methionine) partially reduce interfacial tension, increase methane solubility, and guide gas molecules for efficient and orderly transport into the bulk phase
| [61] | W. Lee, K. Kim, D. W. Kang, Y. H. Ahn and J. W. Lee, Energy & Fuels 2025, 39, 2508-2520.
https://doi.org/10.1021/acs.energyfuels.4c05572 |
| [62] | D. Y. Atta, B. M. Negash, N. Yekeen, A. D. Habte and A. B. A. Malik, Journal of Petroleum Science and Engineering 2021, 199. https://doi.org/10.1016/j.petrol.2020.108241 |
[61, 62]
. This reduces diffusion resistance of gas into water and greatly enhances mass transfer efficiency. Simultaneously, the hydrophobic side chains of amino acids guide water molecules to form precursors of hydrate cage structures, lowering the nucleation energy barrier for hydrates. This rapidly reduces the interface inhibition effect, enabling swift nucleation. However, this alone does not fully explain the synergistic nucleation promotion effect.
Therefore, we propose that hydrophobic interactions
may occur in the aqueous solution, as shown in
Figure 5. The hydrophobic side chains of the hydrophobic amino acids connect with the hydrophobic groups of GSH, compensating for GSH's inherently unstable and disordered adsorption at the gas-liquid interface due to its strong hydrophilicity and weak hydrophobicity. This effectively extends a long hydrophobic side chain to GSH, rendering it structurally similar to SDS with enhanced amphiphilicity. A stable, ordered interfacial layer can more effectively reduce gas-liquid interfacial tension, increasing methane gas solubility and diffusion rate. The hydrophobic region at the interface can adsorb a large number of methane molecules, raising local gas concentration. This reduces the disturbance of water molecules by GSH's hydrophilic groups in a disordered state, instead guiding water molecules to form precursors for hydrate cage structures, lowering the nucleation energy barrier. Therefore, adding hydrophobic amino acids can synergistically promote nucleation with GSH, significantly reducing the induction time.
However, glycine exhibits hydrophilicity. It competes with GSH for water molecules near the gas-water boundary, forming a hydration shell around its own molecules. This further reduces the number of free water molecules, hindering the formation of structures required for hydrate formation. Consequently, water viscosity continues to increase, and gas diffusion resistance into the aqueous phase rises. Thus, glycine enhances GSH's interfacial suppression effect, substantially prolonging the induction time.
Figure 5. In the left panel, GSH, due to its strong hydrophilicity, tends to dissolve in the aqueous phase. However, possessing partial amphiphilicity, some GSH molecules continuously adsorb and desorb at the gas-liquid interface. This creates a dynamic and unstable GSH film. In the right panel, methionine connects its hydrophobic side chain to GSH through hydrophobic interactions, enhancing GSH's hydrophobicity. This enables GSH to form a stable, ordered film at the gas-liquid interface. It significantly reduces interfacial tension, decreases gas diffusion resistance, and promotes hydrate growth.
As shown in
Figure 4(b), the methane adsorption capacity (S) of the amino acid-GSH composite systems exhibited a marked increase compared to the corresponding amino acids alone. However, only the methionine composite system (S = 53.4422) surpassed the value obtained from the GSH (2000 ppm) experiment alone, demonstrating a pronounced synergistic promotion of hydrate growth. The aforementioned theory remains applicable. Incorporating GSH into amino acids disperses a large number of GSH molecules in the solution, providing abundant methane adsorption sites. This partially reduces gas diffusion resistance, significantly promotes hydrate growth, and increases gas storage capacity. Furthermore, the hydrophobic side chains of hydrophobic amino acids form hydrophobic interactions with GSH's hydrophobic groups. This compensates for GSH's inherently unstable adsorption at the gas-liquid interface—a consequence of its strong hydrophilicity coupled with relatively weak hydrophobicity. Similar to SDS, this approach may guide the formation of porous, non-compact hydrate surfaces. This significantly reduces interfacial tension between gas and liquid, generates numerous microbubbles, and increases the gas-water contact surface area. Consequently, gas molecules are efficiently supplied to the water, enabling more thorough hydrate growth. Simultaneously, it provides a greater number of gas adsorption sites, enabling uniform hydrate growth and preventing blockage. Therefore, adding hydrophobic amino acids can synergistically promote growth with GSH, substantially increasing gas absorption capacity, with methionine demonstrating superior efficacy.
Given that the methionine composite system exhibits the most favorable effects in both nucleation induction time and growth gas absorption capacity enhancement, subsequent experiments will continue using the methionine system.
3.3. Effect of GSH Addition on TBAB-Promoted Hydrate Formation
Just as thermodynamic promoters participate in constructing methane hydrate cage structures and occupy a portion of the hydrate cages, TBAB induces a unique semi-cage structure in hydrates, with the tetrabutylammonium cation (TBA
+) occupying four larger pores
. This significantly reduces TBAB's maximum gas storage density. Therefore, for natural gas storage and transportation applications, enhancing gas storage density is more critical than the nucleation induction time difference when the latter is relatively minor. Under current experimental conditions (6.0 MPa, 1°C), as shown in
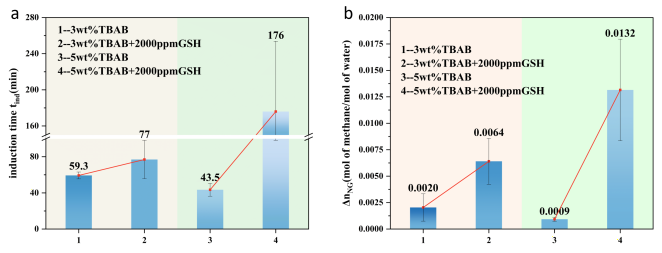
Figure 6, 2000 ppm GSH was added to both 3 wt% and 5 wt% TBAB solutions. Methane hydrate formation tests were conducted before and after GSH addition. We observed that the induction time was extended to varying degrees in the GSH-added systems, with a substantial increase in standard gas absorption (
). The most pronounced change occurred in the 5wt% TBAB system, where induction time increased by 132.5 min and
rose by 1366.7%. This seemingly contradictory phenomenon stems from differing control mechanisms underlying nucleation and growth kinetics.
Figure 6. Adding 2000 ppm GSH to TBAB solutions of varying mass fractions prolongs the induction time and reduces the standard gas absorption. The effect is more pronounced in the 5 wt% TBAB solution, as its higher dosage enhances surface activity, making it more susceptible to GSH influence. Simultaneously, TBAB provides more gas adsorption sites, thereby increasing the standard gas absorption capacity.
Upon GSH addition, its mechanism of action resembles that described in Section 3.1. However, the presence of TBAB introduces distinct phenomena. TBAB, a cationic surfactant, preferentially disperses at the gas-liquid interface in solution. When molecular concentrations are high, the limited adsorption sites at the interface become completely saturated. Thus, GSH competes with TBAB for adsorption sites, reducing TBAB's distribution at the interface and diminishing its ability to adsorb methane molecules. Simultaneously, GSH generates dynamic and disordered interface regions, competing for water molecules near the interface to form hydration shells. This disrupts TBAB's process of arranging water molecules in an ordered manner, severely hindering nucleation and prolonging the induction time.
However, the abundance of TBAB molecules in the aqueous phase far exceeds the relatively scarce GSH molecules, making the effect of increasing nucleation sites negligible. The inhibition of interfacial nucleation thus exerts a greater influence. After the nucleation stage, the growth process of semi-cage hydrates becomes more favorable. The interfacial region, adsorbed by both GSH and TBAB, exhibits a non-compact GSH film due to GSH's strong hydrophilicity, which continuously undergoes adsorption and desorption. This prevents a significant increase in resistance to gas diffusion into the aqueous phase. This mixed GSH-TBAB interfacial layer, unlike pure TBAB, Although pure TBAB hydrate membranes are more porous than pure methane hydrate membranes, they still hinder interfacial mass transfer efficiency. In the mixed case, after nucleation, Instead of forming a dense semi-cage hydrate membrane, a highly porous structure develops that does not impede gas diffusion into water. Hydrate growth can proceed more thoroughly, ultimately increasing methane absorption and significantly enhancing the .
Furthermore, at 5 wt% TBAB, increasing TBAB concentration enhances its surface activity and increases the number of TBAB molecules in the aqueous phase. Stronger surface activity leads to more pronounced disturbance effects upon GSH addition, prolonging the induction time. A higher concentration of TBAB molecules in water provides more gas adsorption sites, enhancing overall gas storage capacity. Upon GSH addition to form a loose, porous interfacial hydrate film, more thorough hydrate formation occurs, releasing greater methane absorption capacity and increasing storage volume.
Given the emphasis on storage density in natural gas transportation research, the 5wt% TBAB system exhibits a larger . Subsequent experiments therefore selected the 5wt% TBAB formulation.
3.4. Effect of Saline Environment on Hydrate Formation in Different Reagent Systems
Freshwater is the source of life, yet Earth's freshwater resources are extremely scarce. Freshwater resources should be prioritized for more critical applications, such as drinking water and agricultural irrigation. We strive to utilize non-potable water (brine) for hydrate formation to store and transport natural gas. Therefore, it is essential to investigate the kinetics of methane hydrate formation in brine environments to shorten induction time, accelerate growth rate, increase methane storage capacity, and advance the development of hydrate-based natural gas storage and transportation.
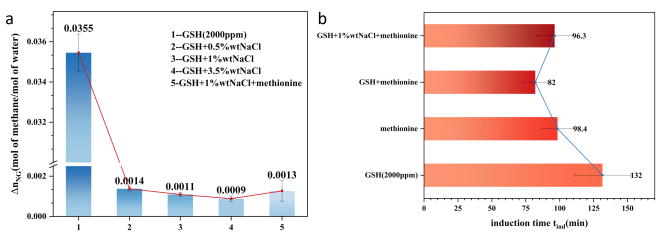
We simulated brine using saltwater solutions containing 0.5wt%, 1wt%, and 3.5wt% NaCl, as shown in
Figure 7. Adding 0.5wt% sodium chloride to GSH (2000ppm) caused a steep 96.3% drop in standard methane absorption (
). Subsequently increasing brine concentration resulted in only a slight decrease (
). Adding methionine (1000 ppm) to a solution containing GSH and 1 wt% NaCl resulted in an almost unchanged
. However, the induction time was significantly reduced, even shorter than the values obtained when GSH or methionine was tested alone. The theory for promoting nucleation induction time described in Section 3.2 remains applicable here.
Methionine exhibits amphiphilic properties, preferentially dispersing at the gas-water interface. This reduces interfacial tension, increases methane solubility, and facilitates efficient, orderly methane transport into water, thereby decreasing gas diffusion resistance. The hydrophobic side chains of methionine form hydrophobic interactions with the hydrophobic groups of GSH, compensating for GSH's inability to stably and orderly adsorb at the gas-liquid interface. This stable, ordered interfacial layer more effectively reduces gas-liquid surface tension. The hydrophobic region adsorbs large quantities of methane molecules, increasing local gas concentration. It minimizes the disruption of water molecules by GSH's hydrophilic groups in a disordered state, instead guiding water molecules to form precursor hydrate cages. This lowers the nucleation energy barrier, significantly reducing induction time. However, the thermodynamic inhibition of 1 wt% NaCl is too potent
, increasing the equilibrium pressure and reducing the hydrate formation rate. Regardless of how fast nucleation occurs, hydrate growth is fundamentally suppressed, resulting in no significant change in the
.
Figure 7. (a) Adding different mass fractions of NaCl to a 2000 ppm GSH solution to simulate a saline environment. Adding just 0.5 wt% NaCl significantly inhibits hydrate formation and growth, with a sharp decrease in standard gas absorption. Adding methionine only slightly increases standard gas absorption, while NaCl's strong inhibition of hydrate growth persists. However, (b) methionine significantly shortens the induction time, even below values observed with GSH 2000 ppm or methionine alone, indicating synergistic promotion remains present.
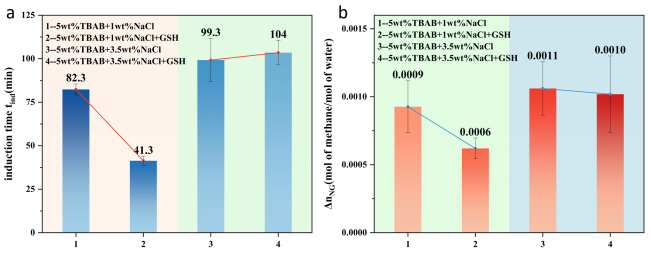
Subsequently, we simulated brine effects by adding 1 wt% and 3.5 wt% NaCl solutions to 5 wt% TBAB. As shown in
Figure 8, adding GSH (2000 ppm) to the solution significantly reduced the induction time in the 1 wt% NaCl system, while
showed only a slight decrease, making it difficult to rule out random effects. In contrast, the induction time for the 3.5wt% NaCl system and
remained virtually unchanged.
We propose that a " salting-out effects " occurred in the 1wt% NaCl system: high-concentration salt ions competed for water molecules, reducing the solubility of GSH's hydrophobic moiety and amplifying its hydrophobicity. This led to spontaneous aggregation into numerous hydrophobic aggregates. These aggregates, similar to micelles with hydrophobic interiors, provided abundant nucleation sites, enriching methane gas, rapidly increasing local gas concentration, and promoting hydrate nucleation. However, primarily due to the increased phase equilibrium pressure at 1 wt% NaCl, the hydrate formation fugacity decreased, with showing only minor changes. Furthermore, at 3.5 wt%NaCl system may exhibit excessive "salting-out effects" due to the high brine concentration, causing GSH to form larger, disordered aggregates. These aggregates cannot effectively diffuse and adsorb at the two-phase interface, losing their nucleation-promoting role. Simultaneously, 3.5 wt% NaCl further deteriorates the thermodynamic environment, drastically reducing the fugacity. Therefore, the induction time and standard gas absorption amount in the 3.5 wt% NaCl system remain virtually unchanged.
Moreover, the induction time for the 1 wt% NaCl system was even shorter than that for the 5 wt% TBAB system in
Figure 6(a). This occurred because the interfacial region became saturated with TBA
+ ions, whose electrostatic repulsion prevented efficient packing. Simultaneously, TBA
+ ions and their hydration shells disrupt the ordered arrangement of water molecules at the interface, prolonging the induction time to some extent. Conversely, 1 wt% NaCl induces a "salting-out effect," displacing TBA
+ ions from the aqueous phase and promoting their compact packing at the interface. This enhances interface order, thereby reducing the induction time. Of course, the applicability of these mechanisms requires further characterization for verification. Subsequent work may employ characterization techniques such as Raman spectroscopy to conduct in situ experiments on methane hydrate formation, monitoring O-H bond changes to detect hydrate nucleation and formation processes.